Right aortic arch: what is it, causes, development options, diagnosis, treatment, when is it dangerous?
The right aortic arch in the fetus is a congenital heart defect, which can occur alone or be combined with other, sometimes severe, defects.
In any case, during the formation of the right arch, disturbances in the normal development of the fetal heart occur. The aorta is the largest vessel in the human body, the function of which is to move blood from the heart to other arterial trunks, up to the arteries and capillaries of the whole body.
Phylogenetically, the development of the aorta undergoes complex changes during evolution. Thus, the formation of the aorta as an integral vessel occurs only in vertebrates, in particular in fish (two-chamber heart), amphibians (two-chamber heart with an incomplete septum), reptiles (three-chamber heart), birds and mammals (four-chamber heart). However, all vertebrates have an aorta, into which arterial blood mixed with venous, or entirely arterial, flows.
During the process of individual development of the embryo (ontogenesis), the formation of the aorta undergoes changes as complex as the heart itself. Starting from the first two weeks of embryo development, there is an increased convergence of the arterial trunk and the venous sinus, located in the cervical part of the embryo, which subsequently migrated more medially, towards the future thoracic cavity. The arterial trunk gives rise not only to two ventricles subsequently, but also to six branchial (arterial) arches (six on each side), which, as they develop, within 3-4 weeks, are formed as follows:
- the first and second aortic arches are reduced,
- the third arch gives rise to the internal carotid arteries that supply the brain,
- the fourth arch gives rise to the aortic arch and the so-called “right” part,
- the fifth arc is reduced,
- the sixth arch gives rise to the pulmonary trunk and the arterial (Botallov) duct.
The heart becomes completely four-chambered, with a clear division of the cardiac vessels into the aorta and pulmonary trunk, by the sixth week of development. A 6-week embryo has a fully formed, beating heart with large vessels.
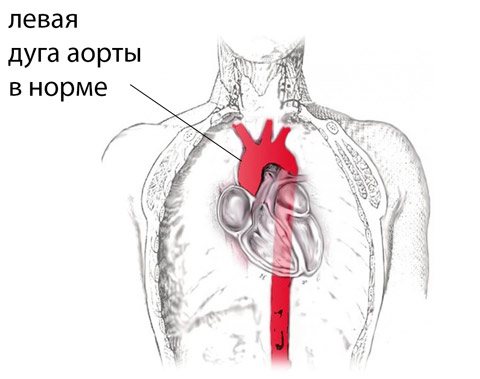
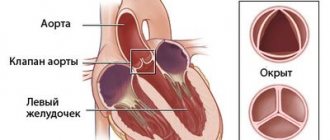
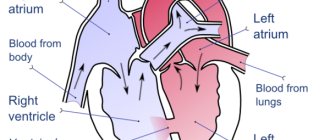
After the formation of the aorta and other internal organs, the topography of the vessel looks like this. Normally, the left aortic arch begins from the aortic bulb in its ascending part, which, in turn, originates from the left ventricle. That is, the ascending part of the aorta passes into the arch approximately at the level of the second rib on the left, and the arch bends around the left main bronchus, heading posteriorly and to the left. The uppermost part of the aortic arch projects onto the jugular notch just above the upper part of the sternum. The aortic arch goes down to the fourth rib, located to the left of the spine, and then passes into the descending part of the aorta.
In the case when the aortic arch “turns” not to the left, but to the right, due to a failure in the formation of human vessels from the branchial arches of the embryo, they speak of a right-sided aortic arch. In this case, the aortic arch extends through the right main bronchus, and not through the left, as it should normally be.
Any malformation is formed in the fetus if a woman is influenced by negative environmental factors during pregnancy - smoking, alcoholism, drug addiction, ecology and unfavorable background radiation. However, genetic (hereditary) factors play an important role in the development of the child’s heart, as well as existing chronic diseases in the mother or past infectious diseases, especially in the early stages of pregnancy (influenza, herpes infection, chickenpox, rubella, measles, toxoplasmosis and many others) .
But, in any case, when any of these factors influence a woman in the early stages of pregnancy, the normal processes of ontogenesis (individual development) of the heart and aorta, formed during evolution, are disrupted.
So, in particular, the period of pregnancy of approximately 2-6 weeks is especially vulnerable to the fetal heart, since it is at this time that the formation of the aorta occurs.
What is the abdominal aorta and where is it located?
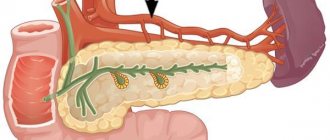
As you know, the largest human artery, the aorta, consists of several sections. Most of them are located within the chest. Only one part (abdominal or abdominal) passes into the abdominal cavity, under the diaphragm. Throughout its entire length, it is located in front of the spine and supplies arterial blood to the entire lower half of the body.
Anatomy of the abdominal aorta
Topographically, this vessel begins at the level of the 12th thoracic vertebra, emerging from the aortic opening of the diaphragm. In the abdominal cavity, the aorta moves anterior to the spinal column, slightly to the left of the midline. Along its entire length, the vessel gives off multiple branches that feed the structures of the abdominal cavity.
The normal dimensions of the abdominal aorta are:
- length – from 13 to 15 cm;
- diameter – 18-20 mm.
The abdominal aorta ends at the level of the 4th or 5th lumbar vertebra, at the point of bifurcation (i.e., bifurcation), where it diverges into the right and left iliac arteries.
Behind the abdominal aorta is the spine, in front is the root of the mesentery of the small intestine, the pancreas and duodenum. On the right is the inferior vena cava, and on the left are the left adrenal gland and kidney.
The branches of the abdominal region are divided into parietal (supplying the abdominal wall) and visceral (supplying the internal organs).
The first group includes the following paired arteries:
- lower diaphragmatic;
- lumbar (4 on each side);
- unpaired sacrum.
Visceral branches are paired and unpaired.
Pairs include:
- middle suprarenal;
- renal (renal);
- testicular (in women - ovarian), which supply blood to the genitals.
- the celiac trunk, which gives branches to the liver, stomach, spleen;
- superior and inferior mesenteric, feeding all parts of the intestine.
In the photo you can see the layout of the outgoing branches
:
Classification of the right-sided aortic arch
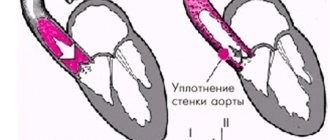
variant of the right aortic arch with the formation of a vascular ring
Depending on the anatomy of the duct anomaly, there are:
- The right aortic arch without the formation of a vascular ring, when the arterial ligament (overgrown arterial, or Botallov, duct, as it should be normally after childbirth) is located behind the esophagus and trachea,
- The right arch of the aorta with the formation of a vascular ring, code arterial ligament, or patent ductus arteriosus, is located on the left of the trachea and esophagus, as if surrounding them.
- As well as a separate similar form, the double arch of the aorta is distinguished - in this case, the vascular ring is formed not by the connective ligament, but by the inflow of the vessel.
Figure: a variety of options for the atypical structure of the aortic arch
Depending on whether any other structures of the heart were damaged during its formation, the following types of defect are distinguished:
- An isolated type of defect, without other developmental anomalies (in this case, if the right-sided aorta is not combined with the DiGeorge syndrome characteristic of it in some cases, the prognosis is as favorable as possible);
- In combination with dextraposition (mirror, right position of the heart and great vessels, including the aorta), (which is also usually not dangerous),
- In combination with a more serious heart defect - in particular tetralogy of Fallot (dextraposition of the aorta, ventricular septal defect, pulmonary stenosis, right ventricular hypertrophy).
Tetralogy of Fallot combined with the right arch is an unfavorable development option
Diagnosis of the defect is not difficult even during pregnancy. This is especially true in cases where the right aortic arch is combined with other, more severe anomalies of heart development. However, to confirm the diagnosis, a pregnant woman is repeatedly examined, including with expert-class ultrasound machines, and a council of geneticists, cardiologists and cardiac surgeons is assembled to make a decision on the prognosis and the possibility of delivery in a specialized perinatal center. This is due to the fact that with some types of defects combined with the right aortic arch, the newborn baby may require heart surgery immediately after delivery.
Regarding the clinical manifestations of the right aortic arch, it should be mentioned that an isolated defect may not manifest itself at all, only sometimes accompanied by frequent obsessive hiccups in a child. In the case of a combination with tetralogy of Fallot, which accompanies the defect in some cases, the clinical manifestations are pronounced and appear in the first days after birth, such as increasing pulmonary heart failure with severe cyanosis (blue discoloration of the skin) in the baby. That is why tetralogy of Fallot is classified as a “blue” heart defect.
What screening shows a defect in pregnant women?
An analysis of fetal DNA can further clarify the absence of a connection between the formation of a right-sided aorta and severe genetic mutations. In this case, chorionic villus material or amniotic fluid is usually collected through a puncture. First of all, DiGeorge syndrome is excluded.
In the event that the right aortic arch is isolated and is not accompanied by any clinical manifestations after the birth of the child, the defect does not require surgical treatment. All you need is a monthly examination by a pediatric cardiologist with regular (every six months - once a year) ultrasound of the heart.
When combined with other heart defects, the type of surgical intervention is selected based on the type of defect. Thus, with tetralogy of Fallot, surgery is indicated in the first year of a child’s life, carried out in stages. At the first stage, palliative (auxiliary) shunts are applied between the aorta and the pulmonary trunk to improve blood flow into the pulmonary circulation. At the second stage, open heart surgery is performed using a cardiopulmonary bypass machine (ACB) to eliminate pulmonary stenosis.
In addition to surgery, cardiotropic drugs that can slow the progression of chronic heart failure (ACE inhibitors, diuretics, etc.) are prescribed for auxiliary purposes.
The prognosis for an isolated right-sided aortic arch is favorable, since in most cases surgical intervention is not even required. So, in general, we can say that an isolated right aortic arch is not life-threatening for the child.
With combined types, the situation is much more complicated, since the prognosis is determined by the type of concomitant heart defect. For example, with tetralogy of Fallot, the prognosis without treatment is extremely unfavorable; unoperated children with this disease usually die in the first year of life. After surgery, the duration and quality of life increase, and the prognosis becomes more favorable.
Source: sosudinfo.ru
Reasons for development
As already mentioned, hypoplasia is the so-called hypotrophy of the aortic vessels, and it entails deformation of the plastic membrane. All this eventually leads to interruption of the aortic isthmus. The reasons for this may be various kinds of hormonal imbalances, pathologies, genetics, and disruptions in innervation. Also, this disease can lead to malfunction of the renal artery.
Similar reasons can lead to coarctation:
- transmission of the disease hereditarily; Negative environmental impact; genetic predisposition in combination with other factors; viral diseases in the first 8 weeks of pregnancy; taking medications in the early stages of pregnancy; drugs and smoking; stress and nervous breakdowns; constant alcohol abuse; regular coffee consumption; working with caustic substances; various diseases of the expectant mother; pregnancy over 35 years of age.
Thus, it is worth saying that hypoplasia should be classified as a rare pathology with non-atherosclerotic and non-inflammatory narrowing of the aorta. Here, mainly only surgical method is used for treatment, which reduces the risk of death or complications. Also, when treating these diseases, reconstruction of the mesenteric and renal arteries must be carried out.
Right aortic arch in the fetus; consequences for the child in the future
How often do they occur, and can they “go away” without surgery?
Congenital heart defects (CHD) are one of the most common developmental anomalies and, according to statistics, occur with a frequency of 7-15 cases per 1000 newborns. In the Krasnoyarsk Territory, in the structure of congenital malformations, malformations of the cardiovascular system of newborns are in first place and account for 42.6% of all defects.
They are the leading cause of infant mortality. Among children born with congenital heart disease, 14-29% die in the first week of life, 19-42% die during the first month, and 30% of infants do not survive one year. CHDs cause up to 50% of all deaths from all developmental defects.
Congenital malformations of the cardiovascular system can be presented in isolated form or be part of a complex of multiple congenital malformations. May be combined with hereditary diseases. More than 40 chromosomal abnormalities have been described that accompany congenital heart disease.
Teratogenic factors that cause congenital malformations, including those of the cardiovascular system, are usually divided into three groups: genetic; exogenous; combined exogenous and genetic (multifactorial genesis).
Genetic teratogenic factors include chromosomal aberrations (numerical, structural), gene mutations that cause developmental defects with a dominant or recessive type of inheritance.
Exogenous factors include viral infections, pharmacological drugs, alcoholic beverages, smoking tobacco products, industrial toxic substances, and so on.
Multifactorial factors include cases of a combined effect when an exogenous factor influences an organism predisposed to a defect. The bulk of congenital diseases, including heart defects, have a multifactorial genesis (50-78%). For the formation of developmental defects, the timing of exposure to teratogenic factors on the developing organs and systems of the fetus is important. For example, the embryonic period from 2 to 8 weeks of pregnancy is considered a critical period of development, it is the most vulnerable to adverse effects on the developing heart and other developing organs of the fetus. Since the developing embryo and fetus are very sensitive to unfavorable factors, they can cause the death of the embryo (fetus), developmental defects from severe, incompatible with life, to mild developmental anomalies, as well as functional disorders that can appear immediately after birth or in further.
Currently, the Krasnoyarsk Regional Medical Genetic Center is engaged in the prevention of hereditary diseases and congenital malformations. The work of the medical genetic center is organized on the basis of decrees of the Government of the Russian Federation and decrees of the Government of the Krasnoyarsk Territory, orders of the Ministry of Health of the Russian Federation, orders of the Ministry of Health of the Krasnoyarsk Territory. Work is being carried out and constantly improved in the following areas: medical and genetic counseling, prenatal diagnosis of child development disorders, screening of newborns for 5 hereditary diseases, clinical examination of families with hereditary pathologies, monitoring of congenital malformations.
Annual monitoring of the outcomes of congenital malformations of the cardiovascular system in newborns and fetuses allows us to assess the number and structure of congenital malformations of the heart in the prenatal period, that is, during the period of intrauterine development of the fetus, and in the neonatal period, as well as to monitor the volume of specialized care was provided, what were the complications and concomitant pathologies.
The opening of the Federal State Budgetary Institution “Federal Center for Cardiovascular Surgery” in Krasnoyarsk allows not only to conduct a comprehensive examination of children with congenital heart defects, prescribe therapeutic treatment and clinical examination according to indications, but also to carry out surgical treatment from the early neonatal period. Today, modern possibilities for surgical interventions are quite large. Experience shows that not all children need intervention immediately when pathology in the heart and main arteries is detected. Some children are not subject to surgery due to the insignificance of anatomical disorders, while others, on the contrary, are unable to correct the defect and severe non-cardiac pathology. To plan care for congenital heart disease, it is important to assess the hemodynamic significance of diseases and the likelihood of developing critical conditions with them. A positive outcome is influenced by early detection of congenital heart pathology in children.
CHD is a group consisting of many nosological forms, and it tends to expand due to the improvement and introduction of diagnostic research methods into medical practice.
Among congenital heart defects, intracardial septal defects are one of the most common congenital heart defects; they can occur with a frequency of up to 60% of all congenital heart defects in fetuses and newborns. They can be hemodynamically significant and, in 90% of cases, predominantly hemodynamically insignificant. There are defects of the interatrial (ASD), interventricular (VSD) and aortopulmonary septa. May be single or multiple. The extreme manifestation of multiple muscle defects is a “Swiss cheese” septum.
In the structure of intracardial septal defects, ventricular septal defects are more common. With an isolated ventricular septal defect, the remaining parts of the heart are developed normally. In the prenatal period, a small (less than 4 mm) VSD does not affect hemodynamics and fetal development. Among the identified defects, small hemodynamically insignificant ones are more often closed by the time of birth and thus do not affect the prognosis for the life and health of the newborn.
The incidence of small, hemodynamically insignificant VSDs in newborns is over 50 cases per 1000 newborns; most of these defects close spontaneously by 1-2 years of life. The process of closing the defect can last up to 10 years. Unfortunately, it is impossible to predict the course of the defect in each specific case.
Currently, an increase in the detection rate of intracardial septal defects among fetuses and “healthy newborns” is associated with echocardiography using color Doppler scanning, which allows the detection of small defects.
VSDs account for up to 20-30% of all isolated congenital heart defects. They have been described in more than 40 chromosomal abnormalities and 90 multiple malformation syndromes.
Clinically significant atrial septal defects (ASD) occur with a frequency of 0.24-0.58 per 1000 newborns, ventricular septal defects (VSD) - with a frequency of 0.38-2.26 per 1000 newborns. The incidence of critical illness in this group of patients may be about 21%. Patients with VSD usually have concomitant cardiac anomalies: ASD, patent ductus arteriosus, coarctation of the aorta, pulmonary or aortic valve stenosis, aortic regurgitation. Indications for surgical intervention are heart failure and delayed physical development in children who are not amenable to therapy.
Such malformations of the cardiovascular system as transposition of the great vessels, tetralogy of Fallot, pulmonary atresia, pulmonary stenosis, hypoplastic left heart syndrome, coarctation of the aorta, interruption of the aortic arch, double exit of the main arteries from the right ventricle, common truncus arteriosus, open ductus arteriosus occur with a frequency of over 30% of all congenital heart defects. These malformations are hemodynamically significant, and the probability of developing critical conditions in children in the first days, weeks, months, and during the first year of life is high: from 30 to 100%. It is clear that the possibility of providing surgical assistance for such pathologies is determined by their detection in the first weeks and months of life.
Prenatal detection of congenital heart defects is facilitated by the development of prenatal diagnostics. The early detection of any congenital malformation of the fetus is based on a well-organized system of screening examinations, including ultrasound, of pregnant women. Ultrasound is the main direct non-invasive method for identifying congenital heart disease and its use in combination with other research methods increases the effectiveness of prenatal diagnosis. Echography differs from other research methods by a combination of such qualities as non-invasiveness, high information content, safety, and the possibility of repeated use in one patient.
During the screening periods of the prenatal period (11 weeks, 3 days - 13 weeks, 6 days; 19 - 21 weeks; 30 - 34 weeks), a comprehensive echocardiographic examination of the fetus is performed. These pregnancy periods are optimal for detecting congenital malformations of the fetus. The use of an expanded research scheme allows you to correctly identify changes in the heart and main arteries, conduct a full differential diagnosis and establish a final prenatal diagnosis of many congenital heart diseases. When detecting congenital heart disease, the risk of chromosomal abnormalities, the risk of combined anomalies, and the risk of non-chromosomal syndromes are taken into account. Prenatal tactics in case of detection of congenital heart disease is that the family of a pregnant woman at a prenatal consultation, which is regularly held on the basis of the Krasnoyarsk Regional Medical Genetic Center, receives complete information from specialists about the nature of the fetal lesion, possible pregnancy outcomes, prognosis for life, quality and health of the unborn child, possible treatment options and makes an informed decision.
Source: angarochka.ru
Ascending aorta
pars
ascendens aortae ,
exits the left ventricle behind the left edge of the sternum at the level of the third intercostal space;
in the initial section it has an extension - the aortic bulb, bulbus aortae .
At the location of the aortic valve, there are three sinuses on the inner side of the aorta,
sinus aortae .
From the beginning of the ascending aorta, the right and left coronary arteries depart.