Arrhythmogenic right ventricular dysplasia (arrhythmogenic right ventricular cardiomyopathy) is a rare disease believed to have genetic causes. It is characterized by structural changes in the structure of the wall of the right ventricle, consisting in the replacement of cardiomyocytes with fibro-fatty tissue, and the development of arrhythmia.
The incidence of arrhythmogenic right ventricular dysplasia (ARVD) ranges from 1 to 6 cases per 10,000 population. It occurs four times more often in men than in women.
After it was established that there is a relationship between sudden cardiac death syndrome and arrhythmogenic right ventricular dysplasia, the interest of scientists in this disease has increased sharply. The results of scientific studies have shown that in children and adolescents under 20 years of age who died from cardiovascular pathology, in every fourth case the myocardium had histological changes characteristic of ARVD.
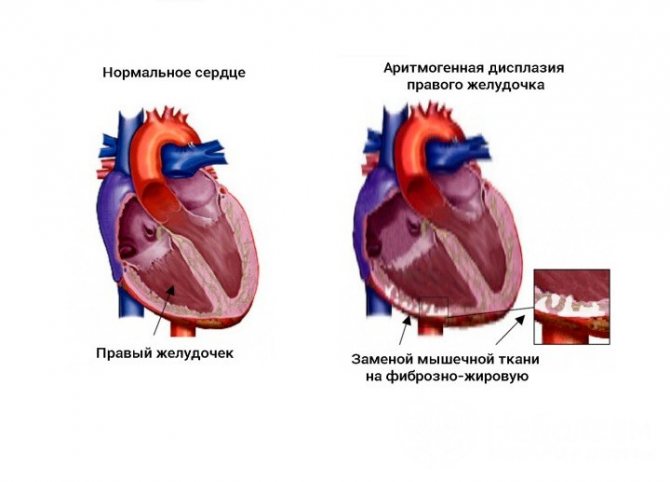
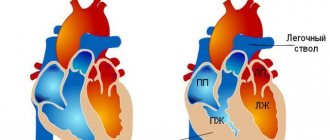
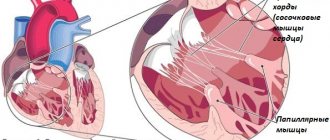
Replacement of cardiomyocytes with fibrofatty tissue is a sign of arrhythmogenic dysplasia of the right ventricle
Causes and risk factors
There is no consensus among the scientific community regarding the reasons underlying the development of arrhythmogenic right ventricular dysplasia. This is largely due to the variety of its clinical forms. It is likely that the pathology includes not one, but several diseases with similar symptoms but different etiologies. However, most researchers believe that genetic mutations lead to the development of ARVD.
Genetic studies in patients with arrhythmogenic right ventricular dysplasia have shown that the latter have abnormalities of genes located on chromosomes 12, 14, 17 and 18. These genes are responsible for the synthesis of specific myocardial proteins. Changes in protein structure lead to a decrease in the resistance of myocardial cells to damaging effects. This causes the myocardial cells to be replaced with adipose tissue over time. An abnormality in the structure of desmoplakin causes a disruption in the transmission of nerve impulses through the myocardium, which leads to the development of arrhythmia.
An important diagnostic sign indicating the presence of possible arrhythmogenic dysplasia of the right ventricle is an indication of the familial nature of the disease, as well as cases of sudden death of relatives.
In addition, there is a form of arrhythmogenic dysplasia of the right ventricle, in which an inflammatory process occurs in the myocardium of the right ventricle, causing the replacement of its cells with fibrous tissue. This form is characterized by a severe course and the transition of the pathological process to other parts of the heart. Presumably, it is also caused by genetic mutations that reduce the resistance of the myocardium to viral damage.
Gene mutations underlying the pathological mechanism of development of arrhythmogenic right ventricular dysplasia are inherited in an autosomal dominant manner with low penetrance, not exceeding 30–50%. This means that even if a child inherits an altered gene from one of the parents, the probability of developing a clinical picture of the disease is no more than 50%.
Scientists suggest that the main cause of ARVD is a genetic mutation
There is only one autosomal recessive inherited form of ARVD with high penetrance (more than 90%) - Naxos disease. It is extremely rare: to date, only 25 cases of this disease are known.
Causes of arrhythmogenic right ventricular cardiomyopathy
At the moment, there is no generally accepted point of view on the causes of the development of AP CMP due to the heterogeneity of the manifestations of the disease. It is possible that AP CMP combines several pathologies with similar manifestations and different etiologies. But the only documented theory to date is hereditary, which explains the occurrence of arrhythmogenic right ventricular cardiomyopathy by a genetic mutation.
When studying the genome of patients with AP CMP, gene abnormalities were identified in the 12th, 14th, 17th and 18th chromosomes - these genes encode myocardial proteins such as alpha-actin, desmoplakin, plakoglobin, plakophyllin and others. Disturbances in the structure of these proteins lead to a decrease in the resistance of cardiomyocytes to damaging factors, which eventually leads to fatty infiltration. However, the main role in the development of arrhythmia during AP CMP is played by dysfunction of the desmosome protein, as a result of which the distribution of excitation throughout the myocardium changes.
In some cases, instead of focal fatty infiltration of the walls of the right ventricle, fibrinous infiltration is observed, which is inflammatory in nature and generally resembles the picture of viral myocarditis caused by the Coxsackie virus, etc. This form tends to spread to the left ventricle and is characterized by a severe course, often leading to the death of the patient . From the point of view of the hereditary theory of the development of AP CMP, it is believed that gene mutations increase the susceptibility of the myocardium to damage by viruses.
Most mutations are inherited in an autosomal dominant manner with a penetrance of 30-50%. One extremely rare form of arrhythmogenic right ventricular cardiomyopathy (Naxos disease - only 25 cases have been described) is autosomal recessive and has a high penetrance of more than 90%. Homozygotes for the mutant gene suffer from malignant ventricular arrhythmia and often die in childhood or adolescence.
Forms of the disease
Depending on the characteristics of inheritance and clinical course of arrhythmogenic dysplasia of the right ventricle, the following forms are distinguished:
- Reference or pure form.
- Naxos disease. It manifests itself as a malignant arrhythmia, which becomes the cause of death in patients in childhood.
- Venetian cardiomyopathy. The wall of the left ventricle is sometimes involved in the pathological process. Most patients die in childhood.
- Smoke disease. Causes sudden cardiac death in adolescent boys.
- Tachycardia without manifestations of heart failure or extrasystoles with a focus of pathological excitation in the right ventricle.
- Extrasystole with a focus of excitation in the right ventricle and the presence of signs of an inflammatory process in the myocardium. Often leads to death.
- Uhl's anomaly. An extremely rare form of ARVD, in which almost all the myocardial cells of the right stomach are gradually replaced by fibro-fatty tissue, which leads to the development of progressive cardiovascular failure and death.
- Non-arrhythmogenic form. In most cases, it is asymptomatic and most often causes sudden cardiac death.
The incidence of arrhythmogenic right ventricular dysplasia (ARVD) ranges from 1 to 6 cases per 10,000 population. It occurs four times more often in men than in women.
Causes and types of cardiomyopathies
Modern classifications try to take into account not only morphological and functional disorders, but also the causes of the disease. Thus, hypertrophic cardiomyopathy is classified as a primary genetically determined pathology (along with arrhythmogenic right ventricular cardiomyopathy, “non-compact myocardium”, “ion channel pathology” and others).
About 125 mutations have been discovered in genes that cause this form of the disease. Among them, defects are most often detected on chromosomes 1, 11, 14, 15. Abnormal cardiac β-myosin is synthesized; troponin T; myosin-associated protein C; tropomyosin; light chain myosin; titin; α-actin; cardiac troponin I; cardiac α-myosin heavy chain.
These anomalies lead to disruption of mutual orientation and chaotic arrangement of cardiomyocyte fibers. The random arrangement of fibers is a morphological substrate for the development of arrhythmia and diastolic dysfunction.
Classification of cardiomyopathy
Since the 60s of the 20th century, attempts have been made to create classifications dividing cardiomyopathies into types based on structural and functional changes in the myocardium.
Most classifications distinguish cardiomyopathy:
- Dilatational. The pathology is characterized by expansion of the cavities of the heart chambers and systolic and diastolic disorders. Severe myocardial hypertrophy is not typical;
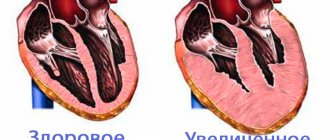
- Hypertrophic. This type is distinguished by significant, often asymmetrical ventricular hypertrophy, accompanied by diastolic dysfunction. No dilatation is noted;
- Restrictive. With restrictive cardiomyopathy, the volume of the ventricles decreases and the diastolic filling of the ventricles worsens. Systolic function is close to normal.
Dilated cardiomyopathy is a mixed type. It is caused by mutations in the genes of sarcomeric proteins, cytoskeleton, nuclear membrane, and mitochondrial cytopathies. In addition to genetic disorders, the role of viral infections and autoimmune reactions has been identified in the pathogenesis.
Normal heart (left) and hypertrophic cardiomyopathy
In 40–50% of patients, cardiomyopathy develops after viral myocarditis. A viral attack destroys cardiomyocytes, the contents of the cells become a target of the immune system. Autoimmune destruction of cardiomycytes that make up the muscle layer is triggered.
Among the non-hereditary causes of this type of cardiomyopathy are also the effects of drugs, alcohol, endocrine disorders, tachycardiomyopathies, eosinophilia and other factors.
Alcoholic cardiomyopathy
Alcoholic cardiomyopathy is one of the main causes of higher mortality in men compared to women. This disease is diagnosed in 86% of cases in middle-aged men. Dyshormonal cardiomyopathy occurs against the background of hormonal therapy, with disorders of estrogenic ovarian function.
Dyshormonal cardiomyopathy
Unlike alcoholic cardiomyopathy, dishormonal cardiomyopathy is more often observed in women, however, men with pathological menopause are also not immune from this disease. This type of pathology can be a consequence of menstrual disorders and gynecological diseases.
Restrictive cardiomyopathy
Restrictive cardiomyopathy is a rare type of pathology, which, like dilated cardiomyopathy, is classified as mixed. Familial (genetically determined) restrictive cardiomyopathy develops with mutations in the genes encoding troponin I, desmin, and HFE gene polymorphism.
Restrictive cardiomyopathy is a mixed type.
Disorders can also be caused by fibrosis, fibroelastosis, thrombosis and be acquired. In particular, this cardiopathy is caused by parasitic infestation, taking certain drugs (anthracyclines), and ionizing effects.
Arrhythmogenic cardiomyopathy
Arrhythmogenic right ventricular cardiomyopathy is diagnosed with a frequency of 1:5000 and often causes sudden death.
The replacement of muscle tissue with adipose and connective tissue in this type of pathology is a consequence of mutations in the genes encoding plakoglobin, desmoplakin, plakofelin and other proteins of the desmosomes of cardiomyocytes.
Symptoms
The clinical course of arrhythmogenic right ventricular dysplasia can vary widely.
The asymptomatic form usually does not manifest itself in any way during the patient’s lifetime. Even with electrocardiography, no changes can be detected.
In the arrhythmic form of ARVD, patients experience tachyarrhythmia and ventricular extrasystole, which are diagnosed during an ECG. Subjective symptoms are usually absent.
ARVD may not manifest itself at all or is characterized by pain in the heart area and tachyarrhythmia
The severe clinical form of arrhythmogenic dysplasia of the right ventricle is characterized by the following features:
- tachyarrhythmia;
- pain in the heart area;
- dizziness.
The most severe course is observed with the development of right ventricular heart failure, which is characterized by the appearance of venous stagnation of blood, edema, including cavitary edema.
Diagnostics
To diagnose arrhythmogenic right ventricular dysplasia, a detailed cardiac examination is performed, including:
- echocardiography;
- electrocardiography;
- X-ray contrast ventriculography;
- magnetic resonance imaging with gadolinium contrast.
If necessary, a puncture biopsy of the myocardium is performed, followed by histological examination of the biopsy sample.
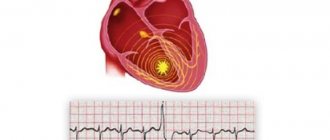
Arrhythmogenic dysplasia of the right ventricle: ECG results
An important diagnostic sign indicating the presence of possible arrhythmogenic dysplasia of the right ventricle is an indication of the familial nature of the disease, as well as cases of sudden death of relatives.
Diagnosis of arrhythmogenic cardiomyopathy
Due to the low prevalence of the disease among the population and the similarity of the signs of the disease with symptoms characteristic of other myocardial pathologies, the clinical examination itself is not very informative. For an accurate diagnosis, long-term monitoring of the patient’s condition and exclusion of more likely heart diseases (myocarditis, other main forms of cardiomyopathy) are necessary.
Diagnostic criteria
Since 1994, the European Society of Cardiology has used a number of diagnostic criteria to identify pathology.
Major diagnostic criteria:
- a marked increase in the volume of the chamber of the right heart and a decrease in the amount of blood released into the vessels during its contraction;
- focal right ventricular aneurysm;
- replacement of the muscular middle layer of the heart with connective and adipose tissue;
- significant segmental dilatation of the right ventricle;
- epsilon wave or local increase (>110 ms) in QRS width in the right precordial leads;
- hereditary nature of the disease (confirmation requires autopsy or biopsy).
Minor criteria:
- mild total dilatation of the right ventricle with an intact left one;
- regional hypokinesia of the right ventricle;
- mild segmental dilatation;
- regional hypokinesia of the pancreas;
- inverted T wave in the right precordial leads (V2, V3) in the absence of right bundle branch block in patients over 12 years of age;
- right bundle branch block in patients over 12 years of age;
- late ventricular potentials;
- ventricular tachycardia with signs of left bundle branch block;
- frequent episodes of ventricular extrasystole (>1000 per 24 hours according to 24-hour monitoring);
- deaths among relatives under the age of 35 years presumably from ACM.
To make a diagnosis, a combination of two major diagnostic criteria or one major and two minor is sufficient. There is an assumption that meeting most of the minor criteria should also be considered as a reason to diagnose the disease in patients whose relatives have previously been diagnosed with ACPM. This makes it possible to detect the disease and begin treatment at an early stage.
Instrumental methods
For diagnosis, the entire range of modern methods for studying heart function is used:
- electrocardiography;
- Holter monitoring;
- chest x-ray;
- endomyocardial biopsy;
- contrast ventriculography;
- magnetic resonance imaging (the most effective method that makes it possible to detect changes such as thinning of the walls and focal aneurysms).
Differential diagnosis
To make a correct diagnosis, it is first necessary to exclude dilated cardiomyopathy (DCM) involving the RV, in which there are signs of its insufficiency, while in ARCM - ventricular arrhythmia. To do this, they resort to endomyocardial biopsy. If the patient suffers from right ventricular cardiomyopathy, when examining the tissues taken, their necrosis, replacement of muscle cells with fat cells, and proliferation of connective tissue are detected. In the case of DCM, noticeable proliferation or partial tissue atrophy and fibrosis in the intermediate layers are found.
Treatment
Conservative treatment of arrhythmogenic right ventricular dysplasia includes the administration of antiarrhythmic drugs. If drug therapy is ineffective, indications for implantation of a pacemaker or cardioverter-defibrillator arise. In case of cardiac right ventricular failure, the patient is prescribed ACE inhibitors.
Timely initiation and regular antiarrhythmic therapy reduces the likelihood of death by 35%.
Surgical methods for treating arrhythmogenic dysplasia of the right ventricle are also used, consisting of excision of pathological foci of excitation followed by suturing of the myocardium. The condition of patients in the first time after surgery improves significantly, but after a few years, 30-40% of operated patients experience a relapse of the disease.
Pacemaker implantation is indicated when drug therapy for ARVD is ineffective
In cases of severe heart failure, the only way to save the patient's life is a heart transplant.
Treatment of arrhythmogenic right ventricular cardiomyopathy
Drug treatment of AP CMP includes antiarrhythmic drugs (amiodarone, sotalol). Reducing the severity of tachyarrhythmias plays an important role in preserving the patient’s life; the cardiologist monitors the effectiveness of drugs using Holter monitoring. In cases where drug therapy is ineffective, they resort to implantation of a cardioverter-defibrillator or pacemaker. For the development of heart failure, ACE inhibitors and carvedilol are used.
Techniques are being developed for the surgical treatment of arrhythmogenic right ventricular cardiomyopathy (ventriculotomy), which boil down to the removal of pathological foci with suturing of the myocardium. The first results of such operations are optimistic, but relapses occur in 30-40% of cases. For severe heart failure, heart transplantation is an effective treatment.
Forecast
Because the disease is highly variable, the prognosis is uncertain. In some patients, 1-2 months pass from the onset of the first symptoms to the development of severe heart failure, while in others, arrhythmogenic right ventricular dysplasia can proceed throughout their lives with minimal or no symptoms.
Timely initiation and regular antiarrhythmic therapy reduces the likelihood of death by 35%. The best results are provided by complex treatment, including drug therapy for arrhythmia and implantation of a pacemaker, in which ventricular fibrillation is practically eliminated.
The development of heart failure significantly worsens the prognosis.
Treatment of cardiomyopathies
Drug treatment of AP CMP includes antiarrhythmic drugs (amiodarone, sotalol). Reducing the severity of tachyarrhythmias plays an important role in preserving the patient’s life; the cardiologist monitors the effectiveness of drugs using Holter monitoring. In cases where drug therapy is ineffective, they resort to implantation of a cardioverter-defibrillator or pacemaker. For the development of heart failure, ACE inhibitors and carvedilol are used.
Techniques are being developed for the surgical treatment of arrhythmogenic right ventricular cardiomyopathy (ventriculotomy), which boil down to the removal of pathological foci with suturing of the myocardium. The first results of such operations are optimistic, but relapses occur in 30-40% of cases. For severe heart failure, heart transplantation is an effective treatment.
Treatment is aimed at correcting severe ventricular arrhythmias and congestive chronic cardiovascular failure. Amiodarone, flecainide, and sotalol are effective in the treatment of ventricular arrhythmias. Amiodarone is prescribed orally at a loading dose of 5-15 mg/kg per day for 10-14 days under electrocardiography control (at saturation, the QT interval increases by 10-15%).
Sotalol is a non-selective β-blocker that is quite effective for the treatment of ventricular tachycardia. It is prescribed to dogs orally 2 times a day at an initial dose of 2 mg/kg and gradually increase it to 6-8 mg/kg per day.
In the presence of congestive chronic cardiovascular failure, its correction is carried out according to well-known principles. ACE inhibitors and carvedilol are especially effective.
There is no specific therapy for cardiomyopathies, therefore all therapeutic measures are aimed at preventing complications incompatible with life. Treatment of cardiomyopathies in the stable phase is outpatient, with the participation of a cardiologist; periodic planned hospitalization in the cardiology department is indicated for patients with severe heart failure, emergency - in cases of development of intractable paroxysms of tachycardia, ventricular extrasystole, atrial fibrillation, thromboembolism, pulmonary edema.
Patients with cardiomyopathies need lifestyle changes:
- decreased physical activity
- following a diet with limited consumption of animal fats and salt
- exclusion of harmful environmental factors and habits.
These measures significantly reduce the load on the heart muscle and slow the progression of heart failure.
For cardiomyopathies, it is advisable to prescribe drug therapy:
- diuretics to reduce pulmonary and systemic venous congestion
- cardiac glycosides for disorders of contractility and pumping function of the myocardium
- antiarrhythmic drugs to correct heart rhythm
- anticoagulants and antiplatelet agents to prevent thromboembolic complications.
In extremely severe cases, surgical treatment of cardiomyopathies is performed: septal myotomy (resection of a hypertrophied area of the interventricular septum) with mitral valve replacement or heart transplantation.