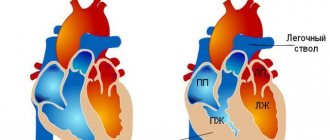
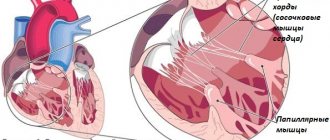
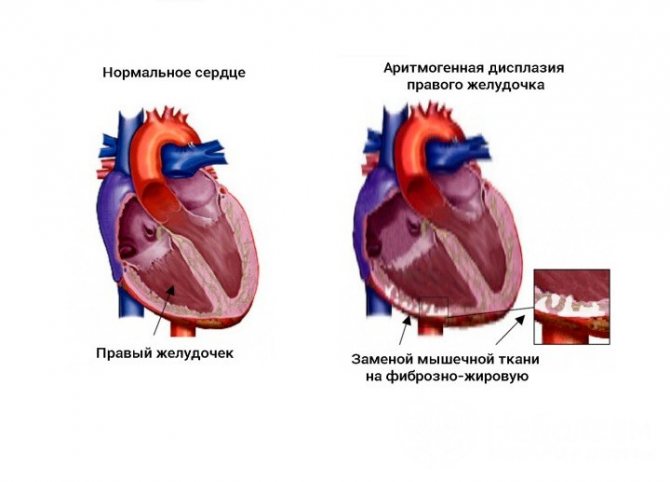
Аритмогенная дисплазия правого желудочка (аритмогенная правожелудочковая кардиомиопатия) – редко встречающееся заболевание, в основе которого предположительно лежат генетические причины. Для него характерны структурные изменения в строении стенки правого желудочка, заключающиеся в замещении кардиомиоцитов фиброзно-жировой тканью, и развитие аритмии.
Заболеваемость аритмогенной дисплазией правого желудочка (АДПЖ) составляет от 1 до 6 случаев на 10 000 населения. У мужчин она возникает в четыре раза чаще, чем у женщин.
После того, как было установлено, что между синдромом внезапной сердечной смерти и аритмогенной дисплазией правого желудочка существует взаимосвязь, интерес ученых к данному заболеванию резко возрос. Результаты научных исследований показали, что у детей и подростков до 20 лет, умерших от сердечно-сосудистой патологии, в каждом четвертом случае в миокарде имелись характерные для АДПЖ гистологические изменения.
Замещение кардиомиоцитов фиброзно-жировой тканью – признак аритмогенной дисплазии правого желудочка
Причины и факторы риска
Среди научного сообщества нет единой точки зрения относительно причин, лежащих в основе развития аритмогенной дисплазии правого желудочка. Во многом это объясняется многообразием ее клинических форм. Вполне вероятно, что патология включает в себя не одно, а несколько заболеваний со схожей симптоматикой, но разной этиологией. Однако большинство исследователей полагают, что к развитию АДПЖ приводят генетические мутации.
Генетические исследования у пациентов с аритмогенной дисплазией правого желудочка показали, что у последних имеются аномалии генов, расположенных в 12, 14, 17 и 18-й хромосомах. Названные гены ответственны за синтез специфических белков миокарда. Изменения структуры белков ведут к снижению устойчивости клеток миокарда к повреждающему воздействию. Это становится причиной замещения со временем клеток миокарда жировой тканью. Аномалия строения десмоплакина вызывает нарушение прохождения нервного импульса по миокарду, что влечет развитие аритмии.
Важный диагностический признак, свидетельствующий о наличии возможной аритмогенной дисплазии правого желудочка, – указание на семейный характер заболевания, а также на случаи внезапной смерти родственников.
Кроме того, существует форма аритмогенной дисплазии правого желудочка, при которой в миокарде правого желудочка возникает воспалительный процесс, обусловливающий замещение его клеток фиброзной тканью. Указанная форма отличается тяжелым течением и переходом патологического процесса на другие отделы сердца. Предположительно, ее причиной также являются генетические мутации, уменьшающие устойчивость миокарда к вирусным повреждениям.
Мутации генов, лежащие в основе патологического механизма развития аритмогенной дисплазии правого желудочка, наследуются по аутосомно-доминантному типу с низкой пенетрантностью, не превышающей 30–50%. Это означает, что даже если ребенок унаследует измененный ген от одного из родителей, то вероятность появления у него клинической картины заболевания составляет не более 50%.
Ученые предполагают, что основной причиной развития АДПЖ является генетическая мутация
Существует только одна наследуемая по аутосомно-рецессивному типу с высокой пенетрантностью (более 90%) форма АДПЖ – болезнь Наксоса. Она встречается крайне редко: на сегодняшний день известно только 25 случаев названного заболевания.
Причины аритмогенной правожелудочковой кардиомиопатии
В настоящий момент общепризнанной точки зрения на причины развития АП КМП нет ввиду гетерогенности проявлений заболевания. Возможно, АП КМП объединяет в себе несколько схожих по проявлениям патологий с различной этиологией. Но единственной документально подтвержденной теорией на сегодняшний день является наследственная, объясняющая возникновение аритмогенной правожелудочковой кардиомиопатии генетической мутацией.
При изучении генома больных АП КМП были выявлены аномалии генов в 12-й, 14-й, 17-й и 18-й хромосомах – указанные гены кодируют такие белки миокарда как альфа-актин, десмоплакин, плакоглобин, плакофиллин и другие. Нарушения структуры этих белков ведут к понижению устойчивости кардиомиоцитов к повреждающим факторам, что и приводит со временем к жировой инфильтрации. Однако главную роль в развитии аритмиипри АП КМП играет нарушение функций белка десмосом, в результате чего распространение возбуждения по миокарду изменяется.
В некоторых случаях вместо очаговой жировой инфильтрации стенок правого желудочка наблюдается фибринозная, имеющая воспалительный характер и в целом напоминающая картину при вирусном миокардите, вызванном вирусом Коксаки и др. Такая форма имеет тенденцию к распространению на левый желудочек и характеризуется тяжелым течением, часто приводящим к смерти больного. С точки зрения наследственной теории развития АП КМП, считается, что мутации генов повышают предрасположенность миокарда к поражению вирусами.
Большинство мутаций наследуются по аутосомно-доминантному типу с пенетрантностью 30-50%. Одна крайне редкая форма аритмогенной правожелудочковой кардиомиопатии (болезнь Наксоса – описано всего 25 случаев) имеет аутосомно-рецессивный характер и высокую пенетрантность – более 90%. Гомозиготы по мутантному гену страдают от злокачественной желудочковой аритмии и часто умирают в детстве или подростковом возрасте.
Формы заболевания
В зависимости от особенностей наследования и клинического течения аритмогенной дисплазии правого желудочка выделяют следующие ее формы:
- Эталонная или чистая форма.
- Болезнь Наксоса. Проявляется злокачественной аритмией, становящейся причиной смерти пациентов в детском возрасте.
- Венецианская кардиомиопатия. В патологический процесс иногда вовлекается и стенка левого желудочка. Большинство больных погибает в детстве.
- Болезнь Покури. Приводит к внезапной сердечной смерти мальчиков-подростков.
- Тахикардия без проявлений сердечной недостаточности или экстрасистол с очагом патологического возбуждения в правом желудочке.
- Экстрасистолия с очагом возбуждения в правом желудочке и наличием признаков воспалительного процесса в миокарде. Нередко приводит к летальному исходу.
- Аномалия Уля. Крайне редко встречающаяся форма АДПЖ, при которой практически все клетки миокарда правого желудка постепенно замещаются фиброзно-жировой тканью, что влечет развитие прогрессирующей сердечно-сосудистой недостаточности и гибель.
- Неаритмогенная форма. В большинстве случаев протекает бессимптомно, чаще всего становится причиной внезапной сердечной смерти.
Заболеваемость аритмогенной дисплазией правого желудочка (АДПЖ) составляет от 1 до 6 случаев на 10 000 населения. У мужчин она возникает в четыре раза чаще, чем у женщин.
Причины и виды кардиомиопатий
Современные классификации пытаются учитывать не только морфологические и функциональные нарушения, но и причины заболевания. Так, кардиомиопатия гипертрофическая отнесена к первичным генетически обусловленным патологиям (наряду с аритмогенной правожелудочковой кардиомиопатией, «некомпактным миокардом», «патологией ионных каналов» и другими).
Обнаружено около 125 мутаций в генах обуславливающие эту форму заболевания. Среди них чаще всего выявляются дефекты хромосомах 1, 11, 14, 15. Синтезируются аномальные кардиальный β-миозин; тропонин Т; связанный с миозином протеин С; тропомиозин; миозин легких цепей; титин; α-актин; сердечный тропонин I; тяжелая цепь кардиального α-миозина.
Эти аномалии ведут к нарушению взаимной ориентации и хаотическому расположению волокон кардиомиоцитов. Беспорядочное расположение волокон является морфологическим субстратом для развития аритмии, диастолической дисфункции.
Классификация кардиомипатии
С 60-х годов XX века делаются попытки составить классификации, разделяющие кардиомиопатии на виды на основании структурных и функциональных изменений миокарда.
Большинство классификаций выделяют кардиомиопатию:
- Дилатационную. Патология характеризуется расширением полостей сердечных камер, систолодиастолическими нарушениями. Выраженная гипертрофия миокарда не характерна;
- Гипертрофическую. Данный вид отличает значительная нередко асимметричная желудочковая гипертрофия, сопровождающаяся диастолической дисфункцией. Дилатация не отмечается;
- Рестриктивную. При кардиомиопатии рестриктивной уменьшается объем и ухудшается диастолическое наполнение желудочков. Систолическая функция близка к норме.
Кардиомиопатия дилатационная относится к смешанным типам. Ее обуславливают мутации в генах саркомерных белков, цитоскелета, ядерной мембраны, митохондриальные цитопатии. В патогенезе помимо генетических нарушений выявлена роль вирусных инфекций и аутоиммунных реакций.
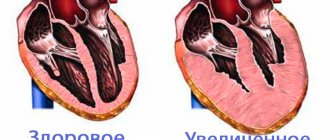
Нормальное сердце (слева) и гипертрофическая кардиомиопатия
У 40–50% пациентов кардиомиопатия развивается после миокардита вирусной природы. Вирусная атака разрушает кардиомиоциты, содержимое клеток становится мишенью иммунной системы. Запускается аутоиммунная деструкция кардиомицитов, составляющих мышечный слой.
Среди ненаследственных причин данного типа кардиомиопатии выделяют также воздействие лекарств, алкоголя, эндокринные нарушения, тахикардиомиопатии, эозинофилию и другие факторы.
Алкогольная кардиомиопатия
Кардиомиопатия алкогольная является одной из основных причин более высокой смертности у мужчин по сравнению с женщинами. Это заболевание в 86% случаев диагностируется у мужчин среднего возраста. Дисгормональная кардиомиопатия возникает на фоне гормональной терапии, при расстройствах эстрогенной функции яичников.
Дисгормональная кардиомиопатия
В отличие от алкогольной, кардиомиопатия дисгормональная чаще отмечается у женщин, однако мужчины с патологическим климаксом тоже не застрахованы от этого заболевания. Данный вид патологии может быть следствием расстройств менструального цикла, гинекологических заболеваний.
Рестриктивная кардиомиопатия
Кардиомиопатия рестриктивная — редкий вид патологии, относящийся, как и дилатационная кардиомиопатия, к смешанным. Семейная (генетически обусловленная) рестриктивная кардиомиопатия развивается при мутациях в генах, кодирующих тропонин I, десмин, полиморфизме гена HFE.
Кардиомиопатия рестриктивная относится к смешанным виду.
Нарушения могут быть также вызваны фиброзом, фиброэластозом, тромбозом и иметь приобретенный характер. В частности, данная кардиопатия вызывается паразитарной инвазией, приемом некоторых препаратов (антрациклинов), ионизирующим воздействием.
Аритмогенная кардиомиопатия
Аритмогенная правожелудочковая кардиомиопатия диагностируется с частотой 1:5000 и нередко становится причиной внезапной смерти.
Замещение мышечной ткани на жировую и соединительную при данном виде патологии является следствием мутаций в генах, кодирующих плакоглобин, десмоплакин, плакофелин и другие белки десмосом кардиомиоцитов.
Симптомы
Варианты клинического течения аритмогенной дисплазии правого желудочка могут варьировать в широких пределах.
Бессимптомная форма при жизни пациента обычно никак себя не проявляет. Даже при проведении электрокардиографии каких-либо изменений выявить не удается.
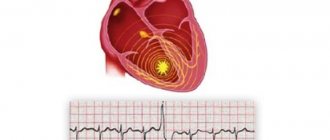
При аритмической форме АДПЖ у больных возникают тахиаритмия, желудочковая экстрасистолия, которые диагностируются в ходе ЭКГ. Субъективные симптомы обычно отсутствуют.
АДПЖ может никак себя не проявлять либо характеризуется болями в области сердца и тахиаритмией
Выраженная клиническая форма аритмогенной дисплазии правого желудочка характеризуется следующими признаками:
- тахиаритмия;
- боли в области сердца;
- головокружение.
Наиболее тяжелое течение наблюдается при развитии правожелудочковой сердечной недостаточности, для которой характерно появление венозного застоя крови, отеков, в том числе полостных.
Диагностика
Для диагностики аритмогенной дисплазии правого желудочка проводится подробное кардиологическое обследование, включающее:
- эхокардиографию;
- электрокардиографию;
- рентгенконтрастную вентрикулографию;
- магнитно-резонансную томографию с контрастированием гадолинием.
При необходимости выполняют пункционную биопсию миокарда с последующим гистологическим исследованием биоптата.
Аритмогенная дисплазия правого желудочка: результаты ЭКГ
Важный диагностический признак, свидетельствующий о наличии возможной аритмогенной дисплазии правого желудочка, – указание на семейный характер заболевания, а также на случаи внезапной смерти родственников.
Диагностика аритмогенной кардиомиопатии
По причине невысокой распространенности заболевания среди населения и схожести признаков болезни с симптомами, характерными для других патологий миокарда, клиническое обследование само по себе малоинформативно. Для точной постановки диагноза необходимо длительное наблюдение за состоянием пациента и исключение более вероятных заболеваний сердца (миокардит, другие основные формы КМП)
Диагностические критерии
Европейское общество кардиологов с 1994 года использует ряд диагностических критериев для выявления патологии.
Большие диагностические критерии:
- выраженное увеличение объема камеры правого сердца и снижение количества выбрасываемой в сосуды крови при его сокращении;
- очаговая аневризма правого желудочка;
- подмена мышечного среднего слоя сердца соединительной и жировой тканями;
- значительная сегментарная дилатация правого желудочка;
- эпсилон-волна или локальное увеличение (>110 мс) ширины QRS в правых прекардиальных отведениях;
- наследственный характер болезни (для подтверждения требуется аутопсия или биопсия).
Малые критерии:
- легкая тотальная дилатация правого желудочка при интактном левом;
- регионарная гипокинезия правого желудочка;
- легкая сегментарная дилатация;
- регионарная гипокинезия ПЖ;
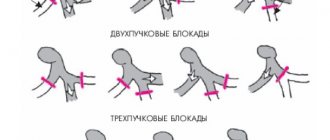
- инвертированный зубец Т в правых грудных отведениях (V2, V3) при отсутствии блокады правой ножки пучка Гиса у пациентов старше 12 лет;
- блокада правой ножки пучка Гиса у пациентов старше 12 лет;
- поздние потенциалы желудочков;
- тахикардия желудочков с признаками блокады левой ножки пучка Гиса;
- частые эпизоды желудочковой экстрасистолии (>1000 за 24 ч согласно данным суточного мониторирования);
- случаи смерти среди родственников в возрасте моложе 35 лет предположительно от АКМП.
Чтобы поставить диагноз, достаточно комбинации двух больших диагностических критериев или одного большого и двух малых. Существует предположение, что соответствие большинству малых критериев также следует рассматривать как повод диагностировать болезнь у пациентов, родственникам которых ранее уже был поставлен диагноз АКПМ. Это позволяет обнаружить заболевание и начать лечение на ранней стадии.
Инструментальные методы
Для диагностики используется весь спектр современных методов исследования работы сердца:
- электрокардиография;
- холтеровское мониторирование;
- рентген органов грудной клетки;
- эндомиокардиальная биопсия;
- контрастная вентрикулография;
- магнитно-резонансная томография (максимально эффективный метод, дающий возможность обнаружить такие изменения, как истончение стенок и очаговые аневризмы).
Дифференциальная диагностика
Для постановки верного диагноза в первую очередь требуется исключить дилатационную КМП (ДКМП) с вовлечением ПЖ, при которой выражены признаки его недостаточности, в то время как при АКМП — аритмия желудочков. Для этого прибегают к эндомиокардиальной биопсии. Если пациент страдает кардиомиопатией правого желудочка, при изучении взятых тканей обнаруживают их некроз, замену мышечных клеток жировыми, разрастание соединительной ткани. В случае с ДКМП находят заметное разрастание или частичную атрофию тканей, фиброз в промежуточных слоях.
Лечение
Консервативное лечение аритмогенной дисплазии правого желудочка включает в себя назначение антиаритмических препаратов. При неэффективности медикаментозной терапии возникают показания к имплантации кардиостимулятора или кардиовертер-дефибриллятора. При сердечной правожелудочковой недостаточности пациенту назначают ингибиторы АПФ.
Своевременно начатая и регулярно проводимая антиаритмическая терапия снижает вероятность летального исхода на 35%.
Применяются и хирургические методы лечения аритмогенной дисплазии правого желудочка, заключающиеся в иссечении патологических очагов возбуждения с последующим ушиванием миокарда. Состояние больных в первое время после оперативного вмешательства значительно улучшается, однако спустя несколько лет у 30-40% прооперированных пациентов возникает рецидив заболевания.
Имплантация кардиостимулятора показана при неэффективности медикаментозной терапии АДПЖ
При выраженной сердечной недостаточности единственный способ спасти жизнь пациента – трансплантация сердца.
Лечение аритмогенной правожелудочковой кардиомиопатии
Медикаментозное лечение АП КМП включает в себя антиаритмические препараты (амиодарон, соталол). Снижение выраженности тахиаритмий играет важную роль в сохранении жизни пациента, контроль эффективности препаратов кардиолог производит при помощи холтеровского мониторинга. В тех случаях, когда медикаментозная терапия малоэффективна, прибегают к имплантации кардиовертер-дефибриллятораили кардиостимулятора. При развитии сердечной недостаточности используют ингибиторы АПФ, карведилол.
Разрабатываются техники хирургического лечения аритмогенной правожелудочковой кардиомиопатии (вентрикулотомия), которые сводятся к удалению патологических очагов с ушиванием миокарда. Первые результаты таких операций оптимистичны, однако рецидивы возникают в 30-40% случаев. При выраженной сердечной недостаточности эффективным методом лечения будет трансплантация сердца.
Прогноз
Так как заболевание отличается высокой вариабельностью, прогноз является неопределенным. У некоторых пациентов от появления первых симптомов до развития тяжелой сердечной недостаточности проходит 1-2 месяца, а у других аритмогенная дисплазия правого желудочка может всю жизнь протекать с минимальными проявлениями или без них.
Своевременно начатая и регулярно проводимая антиаритмическая терапия снижает вероятность летального исхода на 35%. Наилучшие результаты обеспечивает комплексное лечение, включающее медикаментозную терапию аритмии и имплантацию кардиостимулятора, при котором фибрилляция желудочков практически исключается.
Развитие сердечной недостаточности значительно ухудшает прогноз.
Лечение кардиомиопатий
Медикаментозное лечение АП КМП включает в себя антиаритмические препараты (амиодарон, соталол). Снижение выраженности тахиаритмий играет важную роль в сохранении жизни пациента, контроль эффективности препаратов кардиолог производит при помощи холтеровского мониторинга. В тех случаях, когда медикаментозная терапия малоэффективна, прибегают к имплантации кардиовертер-дефибриллятора или кардиостимулятора. При развитии сердечной недостаточности используют ингибиторы АПФ, карведилол.
Разрабатываются техники хирургического лечения аритмогенной правожелудочковой кардиомиопатии (вентрикулотомия), которые сводятся к удалению патологических очагов с ушиванием миокарда. Первые результаты таких операций оптимистичны, однако рецидивы возникают в 30-40% случаев. При выраженной сердечной недостаточности эффективным методом лечения будет трансплантация сердца.
Лечение направлено на коррекцию тяжелых желудочковых аритмий и застойной хронической сердечно-сосудистой недостаточности. В лечении желудочковых аритмий эффективны амиодарон, флекаинид и соталол. Амиодарон назначают преорально в насыщающей дозе 5-15 мг/кг в сутки в течение 10-14 дней под контролем электрокардиографии (при насыщении интервал QT увеличивается на 10-15%).
Соталол — неселективный β-адреноблокатор, достаточно эффективен для лечения желудочковых тахикардий. Назначают собакам перорально 2 раза в сутки в начальной дозе 2 мг/кг и постепенно увеличивают ее до 6-8 мг/кг в сутки.
При наличии застойной хронической сердечно-сосудистой недостаточности проводят ее коррекцию по общеизвестным принципам. Особенно эффективны ингибиторы АПФ и карведилол.
Специфическая терапия кардиомиопатий отсутствует, поэтому все лечебные мероприятия имеют целью предотвращение несовместимых с жизнью осложнений. Лечение кардиомиопатий в стабильной фазе амбулаторное, при участии кардиолога; периодическая плановая госпитализация в отделение кардиологии показана пациентам с тяжелой сердечной недостаточностью, экстренная – в случаях развития некупируемых пароксизмов тахикардии, желудочковой экстрасистолии, мерцательной аритмии, тромбоэмболий, отека легких.
Пациентам с кардиомиопатиями необходимо изменение образа жизни:
- снижение физической активности
- соблюдение диеты с ограниченным потреблением животных жиров и соли
- исключение вредных окружающих факторов и привычек.
Эти мероприятия существенно снижают нагрузку на сердечную мышцу и замедляют прогрессирование сердечной недостаточности.
При кардиомиопатиях целесообразно назначение медикаментозной терапии:
- диуретиков для уменьшения легочного и системного венозного застоя
- сердечных гликозидов при нарушениях сократимости и насосной функции миокарда
- противоаритмических препаратов для коррекции сердечного ритма
- антикоагулянтов и антиагрегантов для предотвращения тромбоэмболических осложнений.
В исключительно тяжелых случаях проводят хирургическое лечение кардиомиопатий: септальную миотомию (резекцию гипертрофированного участка межжелудочковой перегородки) с протезированием митрального клапана либо трансплантацию сердца.