Often children can inherit from their parents not only characteristic traits and positive qualities, but also a hereditary disease.
The likelihood of getting any hereditary disease depends directly on the presence of defective genes in the parents. Thalassemia refers specifically to such genetic diseases that are inherited from parents to their children.
What is thalassemia
Thalassemia is a group of genetic blood diseases (ICD 10 code - D56) that causes abnormal mutation of red blood cells, resulting in a disruption in the transport of oxygen molecules to the cells and tissues of the body.
Hemoglobin makes up the bulk of the blood cell and is the main substance for binding oxygen molecules and moving them throughout the body. With this disease, the entire process of synthesis of hemoglobin polypeptide chains is disrupted, which leads to defective and short-lived red blood cells.
As a result of such changes, there is a lack of oxygen in the tissues of the body, which leads to pathology and deterioration in the functionality of human organs.
The carriers of this disease are defective genes that are responsible for the construction of hemoglobin. Their inferiority lies in the impossibility of compiling the correct set of amino acids in the chromosomal chain.
Causes and biological explanation of the formation of thalassemia
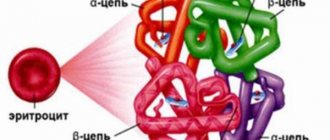
Hemoglobin, as the main culprit in the development of the disease, is a protein of complex structure, which consists of protein and pigment fragments. The protein part of the erythrocyte, in turn, is formed due to the presence of a pair of chromosome sets: beta and alpha sequences. Specific areas called loci in the human gene code are responsible for the correct formation. A locus on the fourteenth chromosome is responsible for the correct evolution of the beta chain, and a locus on the eleventh chromosome is responsible for the formation of the alpha chain.
In thalassemia, pathologies of the chromosome set, due to the loss of one or several segments of the chain, lead to the formation of hemoglobin cells of an erroneous configuration. Protein biosynthesis occurs with significant disruptions, and incorrect coding information at the gene level provokes abnormal assembly of the erythrocyte protein chain in the ribosome. Mutation processes lead to the formation of a protein of “wrong” structure, which, through pathology, cannot fulfill its direct functional duties, namely, supply oxygen to organs and cells, as well as free the body from side components of metabolism. Red blood cells of abnormal origin are characterized by increased fragility; when in contact with the walls of blood vessels and arteries, they have a tendency to self-destruct.
Degradation of red blood cells is accompanied by the release of their constituent components, followed by their sedimentation in the tissues of the body. This process in medicine is called hemosiderosis, while the iron contained in the hemoglobin pigment provokes the destruction of tissues with their further elimination. Tissues that have been destroyed are eventually replaced by connective epithelium, which is unable to perform direct functional tasks, which leads to improper functioning of the affected organ or to its complete failure.
The intensity of pathological processes, as well as the possibility of a patient’s life with such an anomaly, determine the type and extent of the severity of thalassemia.
Forms of thalassemia
Polypeptide chains are responsible for the performance of hemoglobin: alpha (α) and beta (β). Impaired synthesis in any of these chains can result in abnormal red blood cells.
There are two types of this disease based on the irregular abnormality of the polypeptide chains:
- alpha thalassemia (ICD 10 code – D56.0);
- beta thalassemia (ICD 10 code – D56.1).
And also any type of thalassemia may differ in the way the mutant gene is acquired from the parents. Heterozygous thalassemia is acquired from only one parent and is usually a mild form of the disease. Homozygous thalassemia is acquired from both parents and can have a moderate or severe form of the disease.
Alpha thalassemia
Four genes (two genes from each parent) are responsible for building the alpha chain.
The severity of thalassemia disease will depend on the number of transmitted “sick” genes:
- Mutation in one gene. The disease is not detected due to the absence of clinical manifestations. A person with this form of thalassemia can only be a carrier, which will lead to the likelihood of transmitting the disease to their children in the future.
- Mutation in two genes . Thalassemia minor is characterized by a low level of hemoglobin in the blood and small red blood cells. May be acquired from one or both parents.
- Mutation in three genes . The average form of the disease has a clear violation of the oxygen supply of the body. Various pathologies can occur in the systems and organs of the human body.
- Mutation in four genes. Major form of the disease or alpha thalassemia major. With this anomaly, the alpha chains responsible for the transfer of oxygen molecules into cells are completely absent. This form of the disease leads to the death of the child in the womb or in the first days after birth.
Beta thalassemia
Only two genes (one from each parent) are involved in creating the beta chain in hemoglobin. When a mutation occurs in one or two genes of this chain, beta thalassemia appears.
There are three forms of this type of disease:
- Minor or minor form of thalassemia occurs when there is a single mutant gene. It is characterized by a subtle decrease in beta chains, which affects the absence of external signs of the disease. Blood tests reveal small red blood cells. A person with this form of the disease lives a relatively healthy life.
- Intermedia or intermediate form of thalassemia is formed by a slight mutation of two genes. Significant damage to the beta chains occurs, which affects very low hemoglobin levels and the presence of a large number of underdeveloped red blood cells. Without treatment, the moderate form usually becomes severe.
- Major or major form of thalassemia (Cooley's anemia) appears when both genes responsible for beta chains are completely mutated. This form requires constant blood transfusions to maintain hemoglobin levels.
Thalassemia Minor Clinic
The minor or heterozygous form is transmitted by one of the parents. The second “healthy” gene smoothes out damage. Symptoms of thalassemia may not appear for a long time or may cause symptoms common to other diseases:
- weakness, increased fatigue;
- headache;
- dizziness.
During inspection, pay attention to the following:
- pale skin with a jaundiced tint;
- possible enlargement of the liver and spleen.
A child with a minor form has a sharply increased susceptibility to infections and reduced immunity. Viral and intestinal infectious diseases are very severe and frequent.
In adulthood, a woman develops hydrops fetalis during pregnancy (increased intracranial pressure and accumulation of fluid in the ventricles of the brain). This pathology is incompatible with the life of the child; in the case of normal birth, it leads to severe neurological and mental disorders.
β-thalassemia minimalis is called Silvestroni-Bianco syndrome. This form of the disease is practically asymptomatic and is discovered incidentally in families with cases of thalassemia.
Symptoms
In forms of thalassemia minor and major, symptoms manifest differently. According to the method of acquisition, thalassemia minor is classified as heterozygous, that is, the thalassemia gene is transmitted from one parent. In this case, due to the healthy gene from the second parent, the entire overall picture of the disease will be slightly noticeable.
The main symptoms of the minor form include:
- general fatigue, lethargy and weakness;
- frequent headaches and dizziness;
- pallor and yellowness of the skin;
- accumulation of gases, flatulence;
- high susceptibility to viral and intestinal infections;
- possible enlargement of the spleen and liver upon palpation and ultrasound diagnostics.
Moderate and severe forms of thalassemia are classified as homozygous, since defective genes are received from the father and mother at once.
Symptoms for these forms of the disease are pronounced:
- tower-shaped skull;
- formation of Mongoloid facial features;
- enlargement of the upper jaw and expansion of the nasal septum throughout the course of the disease;
- yellowness and pallor of the skin. Also read our article about facial pallor during pregnancy.
- mental and physical developmental delays are observed;
- formation of bone growths on the feet;
- enlargement of the spleen and liver due to lack of oxygen;
Children with severe thalassemia rarely exceed the five-year mark.
During the transition to adolescence, patients experience additional symptoms:
- frequent bone fractures;
- frequent lung infections;
- sepsis due to contact with infection
- delayed sexual development.
Clinical picture
Symptoms
The main symptoms of the disease are:
- yellowness of the skin and mucous membranes (this manifestation is associated with the accumulation of bilirubin in the body, formed during the breakdown of red blood cells);
- pain and ulceration of the skin in the lower extremities;
- gallstone disease caused by the deposition of bilirubin stones in the gallbladder;
- retardation in physical development (patients are short);
- late puberty;
- decreased muscle tone;
- increased fatigue;
- frequent occurrence of infectious diseases (herpetic rashes, acute respiratory and intestinal infections, influenza);
- hydrocephalus (a similar symptom appears during the fetal development stage, when cerebrospinal fluid accumulates in the ventricles of the brain).
Signs
External signs of pathology are different. Their appearance is determined by the form of thalassemia. Children suffering from this genetic disorder are different:
- a special shape of the skull (the bones form a tower structure, which is why the head takes on a square shape);
- flattened bridge of the nose (saddle nose);
- Mongoloid eye shape (the disease causes narrowing of the palpebral fissure);
- protruding upper jaw.
An early sign of thalassemia is an enlarged liver and spleen. This is facilitated by hemosiderosis and impaired hematopoiesis. A large amount of hemosiderin, a pigment released when hemoglobin is destroyed, is deposited in the tissues.
Diagnostics
To identify the disease use:
- Examination of the patient. Helps to identify external signs of the disease and make a preliminary diagnosis.
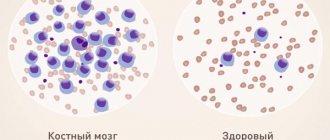
- General blood analysis. A pronounced decrease in hemoglobin levels (up to 30-50 g/l) and the presence of faintly colored red blood cells are detected. The degree of saturation of red blood cells with hemoglobin decreases sharply. The number of reticulocytes—cells from which red blood cells are subsequently formed—increases. An increase in serum iron levels is detected.
- Blood smear. Such a study is aimed at detecting red blood cells that are faintly colored and small in size. The sample contains target cells (red blood cells that have a bright red center and a pale periphery). The cells have different sizes and shapes (oval or sickle-shaped red blood cells are found).
- Biochemical blood test. An increase in the content of unbound bilirubin and iron is detected. The ability of serum to bind iron decreases.
- Electrophoresis of hemoglobin using reagents. Helps determine quantitative indicators of hemoglobin components. With the homozygous form of the pathology, the level of immature hemoglobin in the blood of an adult patient increases.
- Study of hemoglobin synthesis processes in laboratory conditions.
- Bone marrow puncture. The sample obtained during this procedure contains an increased number of sideroblasts (precursors of red blood cells that have large nuclei).
- X-ray of bones. In beta thalassemia, areas of the skull with reduced bone density and hypertrophied areas (with increased thickness and density) are found. In medicine, this is called a hedgehog or brush symptom. The small bones of the lower and upper extremities have transverse striations.
- Molecular analysis. The procedure detects mutations in the beta chains of hemoglobin that interfere with the normal production of components.
- Consultations with a geneticist and hematologist.
Important information: Can there be a high temperature with anemia in adults?
With thalassemia minor, changes in laboratory test results are less pronounced than with the major form of the disease.
Diagnostics
Modern medicine is able to diagnose this blood disease in the early stages if the diagnosis is carried out correctly.
The first step is timely genetic consultation before planning pregnancy to determine the heredity of thalassemia in future parents. Prenatal diagnosis is possible as early as the eleventh week of pregnancy. Early detection of the disease will help to begin to fight thalassemia in advance.
Sometimes a family history and external examination of the patient allows one to suspect a blood disease in the early stages.
To confirm the diagnosis and begin treatment, the following tests are prescribed for thalassemia:
- general blood test - checks the level of hemoglobin, the size and shape of red blood cells, the presence of reticulocytes.
- blood biochemistry - checking bilirubin levels, iron concentration, ability to bind iron.
- PCR – detection of a defective thalassemia gene in the chromosome. Allows you to definitively confirm or reject the diagnosis of thalassemia.
- Ultrasound diagnostics - the size of the liver, spleen and the presence of bilirubin stones are examined.
- bone marrow puncture - the composition and nature of the malfunction of the hematopoietic system.
- X-ray examination - detection of defects of the skeleton and bone tissue.
- specific blood tests to determine damage to the circulatory system.
Specifics of diagnosing the disease
There are a lot of diagnostic methods that can be used to identify hematological diversification in the blood of a patient with thalassemia, which consists of biochemical microanalysis of blood and auxiliary ultrasound studies, supplemented by the patient’s medical history.
The initial diagnosis of thalassemia consists of examining the patient’s blood, which makes it possible to identify the presence of the disease in the early stages, as well as to detect which of the chains of red blood cell formation contains pathology. With thalassemia, a blood test will indicate low hemoglobin parameters, which rarely exceed fifty units. A low color coefficient, elevated reticulocyte count, and high serum iron criteria also indicate the presence of thalassemia. Laboratory blood tests will show distorted, hypochromic, target-shaped or flattened red blood cells.
Visual indicators of the presence of thalassemia may include a change in the patient’s skin color to a jaundiced tint, as well as facial features characteristic of Mongoloid races. When collecting a patient’s life history, it is advisable to pay attention to the patient’s ethnicity, since Latin Americans, Africans and Caucasians are more prone to the disease.
If thalassemia is suspected, the patient is referred for consultation to a hematologist and geneticist to confirm or refute the tentative diagnosis. To verify the diagnosis, the patient may be prescribed an additional ultrasound examination.
Thalassemia in children mainly manifests itself in a very bright form, which allows it to be identified in the early stages and treatment therapy to be started immediately. Modern medicine makes it possible to identify the presence of a disease in the fetus even during its gestation period using a special analysis called screening. In severe forms of the disease in the fetus, thalassemia can be a direct indicator for artificial termination of pregnancy. For people who have a family history of thalassemia patients, or who themselves are carriers of the mutation gene, it is important to visit a geneticist before planning a pregnancy in order to reduce the likelihood of having a child with the pathology.
Causes of the disease
- Thalassemia is the most complex hereditary disease , as it is transmitted from parents to children through defective genes that have undergone mutation. Sometimes parents, being carriers, may not be aware of the existence of this disease in their blood. But thalassemia can fully manifest itself in the genes passed on to children.
- If there are incorrect genes on the chromosomes, hemoglobin synthesis is disrupted, which leads to loss of functionality of red blood cells.
- The outbreaks of this disease occur mainly in the Mediterranean, the Middle East and central Africa, and extremely rarely in Latin America.
- The occurrence of the primary mutation of the parent gene is due to Plasmodium falciparum. It has been scientifically proven that it is Plasmodium, when it enters the human blood, that has the main influence on the mutation of genes in the sixteenth and eleventh pairs of chromosomes, which are responsible for the creation of alpha and beta chains.
Genetic interpretation of beta thalassemia species
Genetic scientists have discovered an interesting pattern: people have the same mutation of the genes responsible for the synthesis of hemoglobin, but the clinical picture and severity of the disease differ.
Genes can be in the following states:
- normal - characteristic of a healthy person;
- partially damaged - “works” incompletely, which is why the synthesis of polypeptide chains is insufficient;
- completely destroyed - the synthesis stops.
On this basis, types of thalassemia are divided into:
- minor - the mildest form, only one gene is damaged, the person looks healthy outwardly, a blood test suggests slight anemia;
- intermedia - the lack of beta chains seriously affects the synthesis, red blood cells are underdeveloped, anemia is expressed with obvious signs, but the body can still adapt, so there is no need for constant blood transfusions;
- major - all genes have undergone mutations, the patient requires constant blood transfusions for health reasons.
Treatment
Thalassemia is still one of the rare treatable diseases. This is primarily due to the poor effectiveness and complexity of the treatment method.
With thalassemia minor, no special treatment is required, but there is an action plan to maintain the patient’s moderate condition:
- Regular collection of blood tests to monitor hemoglobin, red blood cells and iron.
- Compliance with the recommended lifestyle and nutrition.
- Reducing iron levels in the human body using food and medicine.
For moderate and major forms of thalassemia, a number of treatment measures are being developed:
- Constant monitoring of hemoglobin and iron levels in the blood using blood tests.
- Regular blood transfusions to maintain the required number of red blood cells in the blood.
- Elimination of excess iron from the body using a complex of drugs (Desferal).
- If the spleen is greatly enlarged, its resection (removal) is recommended. Not applicable to children under five years of age.
- Bone marrow transplant surgery, which is currently the best treatment option. The complexity of this procedure lies in the individual selection of a donor who meets all the characteristics.
- Take ascorbic acid daily to remove iron from the body.
- Introduction to the diet of foods that reduce the absorption of iron into the body.
Therapy problems
Treatment of thalassemia depends on the severity of damage to erythropoiesis and the degree of gene coverage by mutation processes. The following methods are currently used.
The diet is aimed at reducing the absorption of iron in the intestines; nuts, cocoa, soy, and tea are recommended.
The severe form requires regular blood transfusions, red blood cells, and thawed and filtered red blood cells. The effectiveness is temporary, side effects are possible, but the main thing is to save the patient’s life.
The therapy is supplemented by the daily elimination of excess iron through the administration of chelates - special complexes that increase the effect of the medicine. Desferal is prescribed to bind iron. This drug prevents siderosis (a pathological condition caused by deposition of iron in tissues), but does not affect hemoglobin levels.
If a sharp deterioration of the condition occurs, similar to hemolytic crises, glucocorticoids in large doses are indicated.
Splenectomy is possible for children with a large spleen after the age of five. The most optimal age is 8–10 years. After removal of the spleen, a period of improvement begins, but the risk of infection is dangerous.
For transplantation, a donor matching all parameters is required, preferably a close relative.
Symptomatic remedies include hepatoprotective drugs; large doses of ascorbic acid help remove excess iron from the body.
All forms of thalassemia require medications with folic acid and B vitamins. Against the background of an associated infection, folic acid should be used in large doses during pregnancy, since ineffective hematopoiesis in thalassemia significantly increases its consumption by cells.
What is the danger of the disease
- With a minor form of this disease, the patient is exposed to minimal danger. Since the disease is practically asymptomatic and does not have serious consequences. The only danger is the subsequent transmission of the abnormal thalassemia gene to a future generation.
- With thalassemia major, there is a high mortality rate, especially among newborn patients. In mature and adolescent patients, there is a risk of developing additional serious diseases such as cancer, sepsis, and infections.
Disease prognosis
Thalassemia minor has a favorable prognosis. Dangerous consequences are rare, and most patients do not require ongoing treatment. The intermediate type of the disease also has a good prognosis, but patients require constant blood transfusions and the administration of drugs that bind and remove iron. The high number of deaths from this disease is associated with the deposition of this element in the tissues of internal organs.
With severe thalassemia, most patients rarely survive to adulthood; death can occur in the first months of life.
Thalassemia prognosis
For patients with thalassemia, the prognosis depends on the severity of the disease:
- with thalassemia minor often live full, long lives. The difference between such a patient and a healthy person is weaker immunity to various infections.
- Patients with severe thalassemia have an isolated lifestyle. In newborns with this disease, all symptoms appear in the second year of life. People with thalassemia require regular blood transfusions to keep the body functioning. Often the consequences of thalassemia are diabetes, liver cirrhosis, myocardial infarction, and severe joint diseases.
- When this disease appears in a family history, there is a risk of further spread and infection of thalassemia in all subsequent generations.
What are the symptoms of thalassemia?
Symptoms of thalassemia depend on the type of disease and the severity of the anemia. Some people have either no symptoms at all or minor symptoms. For others, symptoms are moderate or severe. In general, the most common signs of the disease include:
- Pallor
- Fatigue, lack of strength, or muscle weakness (fatigue)
- Dizziness or difficulty breathing
- Lack of appetite
- Dark color of urine
- Bilefulness (yellowing of the skin and whites of the eyes)
- Children have slow development and delayed puberty
- Bone deformities on the face
- Bloating
In children with congenital thalassemia, symptoms may appear immediately or over time. Typically, most symptoms appear during the first two years of life. If you notice that your child is developing slower than his peers, it is very important to find out the possibility of thalassemia, since if not properly treated, this disease can lead to heart failure and infection.
Prevention
The main goal of prevention is to identify the risk factor for inheriting thalassemia.
There is a set of measures aimed at preventing and preventing this disease:
- Prenatal diagnostics and genetic consultation, which identify the predisposition of parents and the unborn child.
- Prenatal diagnosis. It is carried out when an abnormal gene is detected in one or both parents. Sometimes the result of such a diagnosis can be a planned termination of pregnancy.
- Constant monitoring of patients diagnosed with thalassemia by geneticists, hematologists and therapists.
- Patients with thalassemia are required to constantly follow a regimen that helps avoid infections.
Every person who does not know his family history is required to undergo a genetic test in advance to detect thalassemia in the blood. It is important to know that this disease is not acquired during life, the child is already born with thalassemia in his genes.
Signs of thalassemia
This is a disease that belongs to a group of hereditary blood diseases that are usually detected in childhood. It is caused by a violation of the synthesis of hemoglobin, which is vital for the transport of oxygen in the blood. In this article we will talk about what kind of disease thalassemia is - signs, forms, treatment, diagnosis, causes of the disease.
Hemoglobin is a complex molecule that is part of red blood cells and is necessary for the delivery of oxygen to the body's tissues. When heme synthesis is disrupted, anemia develops, and body tissues do not receive enough oxygen for metabolic needs.
Normal hemoglobin is made up of four protein chains called globins, each linked to an oxygen-carrying heme molecule. There are two types of globin - alpha and beta. They join together to form heme molecules. The number of chains is approximately the same, so they are in balance with each other.
In children with thalassemia, the formation of either alpha chains or beta chains is impaired. This leads to the synthesis of defective hemoglobin. It is the lack of normal hemoglobin in the bloodstream that leads to the development of anemia and symptoms of thalassemia disease.
An enlarged spleen is often one of the symptoms of the disease. It traps red blood cells, which leads to a decrease in their number in the blood vessels and, consequently, to increased anemia.
Sometimes removal of the spleen improves the condition of sick children. But patients who have undergone splenectomy. susceptible to infection by pneumococcal bacteria. They need to be vaccinated and given penicillin throughout their lives to prevent infections.
There are two main forms of thalassemia:
- beta, in which the formation of B-globin chains is impaired,
- alpha, in which the formation of aglobin chains is impaired.
Both types are divided into different subtypes of varying severity. In all of its forms, alpha symptoms are usually less severe.
The disease is incurable, so affected children may suffer from its consequences for the rest of their lives. However, the use of a number of treatment methods helps patients cope with it.
Multiple blood transfusions are the mainstay of treatment. Once diagnosed, children receive regular blood transfusions, usually every 4 to 6 weeks. The goal of these procedures is to increase the number of different types of cells in the blood (the number of formed elements in the blood) and thereby reduce the degree of anemia. Blood transfusions are absolutely necessary for patients with beta thalassemia major. Without them, patients will die.
Treatment of thalassemia with blood transfusions corrects the degree of anemia and prevents the development of characteristic bone changes in children.
The main problem that arises with multiple blood transfusions is the formation of excess iron in the body. This can cause damage to the liver, heart and other organs.
- To reduce iron toxicity, infusions of the drug deferoxamine are used.
- Children are prescribed oral vitamin C to further remove iron from the body.
Treatment increases the average life expectancy of patients with beta thalassemia major, but does not bring cure. In some cases, children survive to adulthood. The prognosis is very unfavorable. Despite treatment, children with this type of thalassemia rarely survive to puberty.
Thalassemia – what is it? Causes, symptoms, diagnosis and treatment of thalassemia
Currently, there are a huge number of hereditary diseases that a child receives along with genes from his mother or father. For some to manifest, it is necessary that both parents pass on the defective gene to their child. Thalassemia is one of these diseases. Few people know what kind of disease this is. In our article we will try to figure this out.
What is thalassemia
This is not just one, but a whole group of hereditary blood diseases that have recessive inheritance. That is, the child will receive it if both parents pass on the diseased gene to him. In this case, they say that there is homozygous thalassemia. The disease is characterized by disruption of the production of hemoglobin, which plays a major role in the transport of oxygen throughout the body.
Hemoglobin is a protein that has a protein part and a pigment part. The first consists of polypeptide chains: two alpha and two beta. Failure can occur in any of them, hence alpha thalassemia and beta thalassemia.
Impaired hemoglobin synthesis leads to a reduction in the lifespan of red blood cells, and this entails oxygen starvation of cells and tissues. This process triggers a whole chain of reactions leading to the formation of various pathologies in the body.
Classification of the disease
There are several approaches to classifying this disease. If we consider in which circuit the failure occurred, we can distinguish:
- alpha thalassemia;
- beta thalassemia;
- delta thalassemia.
In each case, the severity of symptoms may vary significantly. Taking this into account, we distinguish:
- light form;
- average;
- heavy.
Depending on whether the child received the gene from both parents or from one, the disease is divided into:
- Homozygous, in this case the diseased gene passes from mom and dad. This form is also called thalassemia major.
- Heterozygous. Inherited from only one of the parents.
All varieties are characterized by their symptoms and severity.
Causes of the disease
Each disease has its own causes; thalassemia is also formed under the influence of genes that the child receives from his parents. This genetic disease is particularly complex, but it is also the most common in the world.
Thalassemia is inherited in a recessive manner through the autosome of the parents. This means that the probability of getting sick is 100% for those who received defective genes for this trait from their mother and father.
The disease develops when a mutation occurs in the genes that are responsible for the synthesis of hemoglobin. The alpha form of this disease is quite common in the Mediterranean and Africa. Some people associate thalassemia with malaria, as these areas often experience outbreaks of the disease.
Malarial plasmodium is blamed for the fact that a mutation occurs in genes and thalassemia develops; the photo demonstrates that a large number of cases are found in Azerbaijan, approximately 10% of the total population. This confirms that the prevalence of the disease is associated with mutations, and climatic conditions also influence the mutation process.
Symptoms of thalassemia major
If a child develops homozygous or thalassemia major, symptoms begin to appear almost immediately after birth. These include:
- The skull is shaped like a tower.
- The face has a Mongoloid appearance.
- The upper jaw is increased in size.
- After some time, expansion of the nasal septum can be noted.
- When diagnosed with thalassemia, a blood test shows hepatomegaly, which will ultimately result in the development of liver cirrhosis and diabetes mellitus. An abnormal blood formula will lead to the deposition of excess iron in the heart muscle, and this can lead to heart failure.
- Due to impaired hemoglobin synthesis, tissues and cells experience constant oxygen starvation, which leads to the appearance of multiple pathologies throughout the body.
- The child is lagging behind both in mental and physical development.
- Closer to the age of one, one can notice the growth of bone tissue in the feet due to the destruction of the cortical layer of the bones.
- Ultrasound shows an enlarged spleen
- Yellowness of the skin.
If a child is diagnosed with thalassemia, the symptoms are pronounced, then there is a high probability that he will not live to see his second birthday.
Symptoms of Thalassemia Minor
When the pathology is inherited from only one of the parents, we can talk about thalassemia minor or heterozygous. Since there is a second healthy gene in the genotype, it significantly smoothes out the manifestation of the disease, and symptoms may not appear at all or give a smoothed picture.
Thalassemia minor has the following main symptoms:
- High and rapid fatigue.
- Reduced performance.
- Frequent dizziness and headache.
- Pale skin with signs of jaundice.
- The spleen may also be enlarged.
Despite the smoothed symptoms, the danger lies in the fact that the body’s susceptibility to all infections greatly increases.
Diagnosis of the disease
Medicine has the opportunity to make a diagnosis of thalassemia at the early stages of development; diagnosis is carried out on the basis of laboratory blood tests. They immediately show that hemoglobin has a disturbed structure. You can even determine which of the chains there are deviations.
In young children, the signs of thalassemia appear quite clearly, so there is usually no difficulty in making such a diagnosis. Parents, before deciding to have a child, should undergo examination, especially if there is a carrier of the gene or a patient in the family.
It is possible to make a diagnosis of thalassemia already in the early stages of pregnancy; amniotic fluid is taken for analysis and examined. It will always contain fetal red blood cells, upon examination of which the presence of pathology can be determined.
Early diagnosis is very important because it is possible to begin treatment without waiting for the birth of the child, which will give the most effective result.
Types of beta thalassemia
The clinical picture of the disease can be different, depending on this, beta thalassemia is divided into several groups. Not everyone is familiar with the concept of thalassemia; that this disease depends on many genetic factors is also not known to everyone.
There are several gene states that control the production of beta chains:
- Normal gene. It is in this state that it is found in all healthy people.
- A gene almost destroyed by mutation. The beta chain is not synthesized at all.
- A partially damaged gene can only partially perform its job, so chain synthesis occurs, but in insufficient quantities.
Taking all this into account, the following types of thalassemia are distinguished:
- Thalassemia minor. A mild form of the disease is formed under the influence of just one damaged gene. According to external indicators, the person is completely healthy. Only blood tests diagnose slight anemia and small red blood cell size.
- Thalassemia intermedia. There is already a serious lack of beta chains. The process of hemoglobin formation is significantly impaired, and underdeveloped red blood cells are also formed. Anemia is already evident, but constant transfusions are not needed. Although over time this form can develop into a more severe form, everything will depend on the body's ability to adapt to the lack of hemoglobin.
- Thalassemia major. The mutation affects all genes responsible for the synthesis of beta chains. This thalassemia (photos of patients can be seen in the article) requires constant blood transfusions to save the patient’s life.
Types of alpha thalassemia
Depending on the degree of gene mutation, this form of the disease is divided into several groups:
- A mutation occurs at one gene locus. In this case, clinical manifestations may not be observed.
- The lesion occurs in two loci, and they can be on the same gene or on different ones. A blood test can easily diagnose low hemoglobin levels and small red blood cells.
- Three gene loci are susceptible to mutation. The transfer of oxygen to tissues and organs is disrupted. In some cases, the spleen also becomes enlarged.
- Mutation in all loci leads to a complete absence of alpha chain synthesis. With this form, fetal death occurs while still inside the mother's womb or immediately after birth.
If you have a mild form of alpha thalassemia, treatment may not be necessary, but with a severe form you will have to be under the supervision of doctors all your life. Only regular courses of therapy can improve a person’s quality of life.
Disease prevention
For doctors and geneticists, if there is a diagnosis of thalassemia, it is clear that it is not curable. We have not yet found ways and methods to cope with this disease. But there are still measures to prevent it. The following preventive measures can be mentioned:
- Carrying out prenatal diagnostics.
- If both parents have this disease, then it is imperative to diagnose the fetus in order to identify this pathology. In some cases, it may be necessary to terminate the pregnancy.
- If you have relatives in your family with this diagnosis, it is advisable to visit a geneticist before planning a pregnancy.
Every organism contains a huge number of mutating genes; it is practically impossible to predict where and when a mutation will appear. This is why genetic consultations exist to help married couples understand their ancestry, or rather, diseases that are passed on from one generation to another.
Thalassemia: causes, 5 common clinical signs and 9 diagnostic methods
Thalassemia is a disease based on a violation of the synthesis of hemoglobin chains. It belongs to quantitative hemoglobinopathies.
Etiology
Thalassemia is caused by point mutations or deletions in the genes encoding hemoglobin chains. As a result, this can lead to a decrease in synthesis or complete absence of one of the chains in the body. The other chain forms inadequate hemoglobin tetramers, which leads to the destruction of red blood cells and hemolytic anemia.
The structure of hemoglobin
Hemoglobin is a protein found in red blood cells that is responsible for transporting oxygen to tissues and carbon dioxide from them.
Hemoglobin (we are talking about HbA) consists of four chains: two alpha subunits and two beta subunits. This hemoglobin makes up 97% of the total content in red blood cells.
The remaining 3% is hemoglobin HbA2, which differs in the structure of its two chains: instead of beta subunits, it has delta subunits. HbA and HbA2 are normal when they are in the correct ratio.
Each of the hemoglobin chains binds to its non-protein part - heme.
So, with thalassemia, the synthesis of one of the hemoglobin chains is disrupted: either alpha or beta. According to this principle, there is a classification of thalassemia into:
- alpha thalassemia;
- beta thalassemia.
Thalassemia is classified according to severity:
- mild;
- moderate;
- severe.
There are physiological and pathological types of hemoglobin.
Hemoglobin, which may be normal in humans, includes:
- HbP – primitive hemoglobin, found in the embryo between the 7th and 12th weeks of life;
- HbF – fetal hemoglobin, contains two alpha and two gamma chains, appears after 12 weeks of intrauterine development, in adults its content is less than 1%;
- HbA – hemoglobin of adults, the proportion is 97%, contains two alpha and two beta chains;
- HbA2 – hemoglobin of adults, the proportion is 2%, contains two alpha and two delta chains,
- HbO2 – oxyhemoglobin, formed when oxygen binds in the lungs;
- HbCO2 – carbohemoglobin, is formed when carbon dioxide binds in tissues.
Pathological forms of hemoglobin include:
- HbS – hemoglobin, determined in sickle cell anemia;
- MetHb – methemoglobin, contains a trivalent iron ion, when normally it is divalent. This form is formed when consuming sulfonamides, nitrates, or vitamin C deficiency. Methemoglobin is not able to bind oxygen, resulting in tissue hypoxia;
- HbCO – carboxyhemoglobin, is formed when there is excess carbon monoxide in the inhaled air. It is present in small concentrations in the blood, but its level may increase depending on the characteristics of the inhaled air.
HbS is a type of hemoglobin that occurs when the beta chain is mutated (one amino acid is replaced by another). It occurs in people with sickle cell anemia. Red blood cells containing such hemoglobin do not live long and are quickly destroyed, which is good in the habitats of the malarial plasmodium. People with sickle cell disease have resistance to this parasite.
Thalassemia is sometimes called Mediterranean anemia. This applies most of all to beta thalassemia, which is most often found in the Mediterranean countries, North Africa, and Western Asia. Alpha thalassemia is common in South Asia and West Africa.
Frequency of occurrence
Thalassemia is inherited in an autosomal recessive manner. This means that it affects both boys and girls equally. The recessive nature suggests that children with thalassemia appear in those families where mom and dad are both carriers of mutations. Although they may not even realize that they are carriers, since the symptoms are sometimes invisible.
The average incidence is 1 in 100,000 people and may vary by region. Beta thalassemia is more common than alpha thalassemia.
There are 4 gene regions that encode the synthesis of the alpha chain of hemoglobin. If there is a genetic defect (mutation), alpha thalassemia occurs in them. There are several forms of alpha thalassemia.
Homozygous
When it occurs, the synthesis of alpha subunits in all four sections of the gene is disrupted. As a result, hemoglobin is formed, consisting entirely of beta chains. It is also called Barth's hemoglobin.
Such hemoglobin has a high affinity for oxygen. This means that it has difficulty delivering oxygen to tissues.
As a result, a lack of this gas occurs, which leads to the development of heart failure, edema, dropsy and, as a consequence, intrauterine fetal death.
H-hemoglobinopathy
H-hemoglobinopathy is a form in which the synthesis of the alpha chain in three gene regions is disrupted. As a result of excess beta subunits, HbH is formed. It also has a fairly high affinity for oxygen, it is not stable, and is easily oxidized.
The presence of such hemoglobin in red blood cells changes the red blood cell membrane and is accompanied by the development of hemolytic anemia, that is, increased breakdown of red blood cells. This form of alpha thalassemia appears by the age of 1 year in the form of hemolytic anemia.
There are signs of anemic syndrome and an enlarged spleen. Blood tests show increased reticulocyte values, which indicates an increased regenerative ability of the bone marrow to restore damaged cells. The red blood cells are hypochromic and target-like.
Alpha thalassemia minor is a form in which the synthesis of 1 or 2 alpha chains is impaired. With this form there are no severe clinical manifestations. A mild degree of anemia (microcytic, hypochromic) may be observed in the blood.
Despite the fact that 1 or 2 genes responsible for the synthesis of alpha subunits are disrupted, this does not lead to such serious disorders as, for example, with beta thalassemia.
Beta thalassemia minor
Beta thalassemia minor (minor) is a heterozygous form characterized by a mild course.
In laboratory tests:
- the number of sideroblasts is normal or increased;
- hypochromic microcytic anemia;
- anisocytosis - red blood cells of different sizes;
- poikilocytosis - red blood cells of different shapes;
- increased number of reticulocytes;
- iron is normal;
- elevated levels of indirect bilirubin.
In patients with beta thalassemia minor, there is an increase in the level of HbA2 (up to 6%, normal is up to 3%) and HbF (up to 7%, normal is less than 1%).
Beta thalassemia minor is often confused with iron deficiency anemia and is mistakenly treated with iron supplements. To differentiate between these two conditions, it is necessary to evaluate the level of iron in the blood. With beta thalassemia it will be within normal limits.
Thalassemia major
Thalassemia major (Cooley's anemia) is homozygous and is considered a severe progressive form of beta thalassemia.
Like other forms of thalassemia, it is manifested by pale skin and an enlarged spleen. They appear by the child's 1st year of age.
A feature of Cooley's anemia is bone changes: protruding cheekbones, narrow palpebral fissures, square skull, flat bridge of the nose. Such children have low physical development.
Laboratory tests indicate:
- increased content of sideroblasts;
- low MCV, MCH, MSHC values, indicating hypochromic microcytic anemia;
- anisocytosis;
- poikilocytosis – in the form of a target, schizocytes;
- basophilic puncture of erythrocytes;
- increased osmotic resistance of red blood cells;
- increase in unconjugated bilirubin;
- excess iron content, up to its deposition in organs (hemosiderosis).
Beta thalassemia major is characterized by an increase in the content of fetal (HbF) hemoglobin in the blood to 70%, which is confirmed by electrophoresis.
Sideroblasts - they are also called erythroblasts. They contain non-hemoglobin iron in the cytoplasm in the form of hemosiderin and ferritin.
- Hemolytic anemia will lead to pallor, lethargy and sometimes even yellowing of the skin .
- The abdomen will be enlarged due to splenomegaly (entrainment of the spleen).
- In some forms of thalassemia, bone deformities .
- Children with thalassemia experience delayed physical development due to a lack of oxygen for tissue growth and development.
- Due to increased absorption of iron in the intestines and frequent blood transfusions, an increase in this element in the blood is observed; it is deposited in organs and tissues (for example, heart, liver), disrupting their functioning.
Diagnosis of thalassemia
When diagnosing thalassemia, one cannot rely only on the clinic alone, especially since it may not always be present, for example, in mild forms. Therefore, for an accurate diagnosis it is necessary to “look” inside the body and evaluate the processes occurring in it. Laboratory blood tests will help with this.
Anemia will be indicated by low levels of hemoglobin and red blood cells . A microscopic examination of the blood smear will reveal the shape of the red blood cells and the hemoglobin content in them. That is, the microcytic hypochromic nature of anemia will be confirmed.
Hemoglobin electrophoresis will allow you to assess how its fractional composition has changed (the ratio of HbA, HbA2, HbF to each other).
An ultrasound examination will show an increase in the size of the spleen.
X-rays are used to study bone deformities.
Bone marrow puncture will allow you to evaluate the processes of hematopoiesis and make a conclusion about the severity of the processes.
The main study that can confirm the presence of thalassemia is molecular genetic analysis . An identified mutation in the alpha or beta chain gene indicates thalassemia.
A biochemical blood test will determine a number of indicators that may increase with thalassemia, for example, indirect bilirubin. Assessing iron metabolism in the body will allow for differential diagnosis between iron deficiency anemia and thalassemia.
Before starting treatment, it is necessary to conduct a genetic study to determine the type of mutation and further prognosis for the development of the disease.
Treatment of thalassemia
Thalassemia is a hereditary disease, so there is only symptomatic therapy.
Treatment for thalassemia will depend on the form. For mild forms, therapy is not carried out. If necessary, blood transfusions . Patients with beta thalassemia need to monitor their blood iron levels and, if they have excess levels, they should be prescribed iron-binding medications .
Patients with severe forms are prescribed blood transfusions in combination with drugs that remove iron from the body. Hemoglobin is maintained at 100 g/l.
Deferoxamine is one of those drugs that can form complexes with iron and remove it in the urine. Do not take anything on your own, only take all medications as prescribed by your doctor.
If symptoms do not improve after a blood transfusion, surgery may be performed to remove the spleen.
What are the possible complications of thalassemia?
- The accumulation of excess iron in organs and tissues, which leads to a malfunction in their functioning.
- After removal of the spleen (an organ of the immune system), patients with thalassemia are prone to infectious diseases.
- Delayed physical development in children.
- The occurrence of heart failure.
Forecast
The prognosis is favorable in the presence of mild forms of thalassemia. Such patients rarely have complications and do not require ongoing therapy. With proper treatment of severe forms, unfavorable outcomes can be avoided.
Conclusion
So, to summarize, what to keep in mind:
- thalassemia is a hereditary disease, severe forms of which can appear in children of the first or second year of life, and mild forms of which can be asymptomatic even in adults;
- laboratory indicators characteristic of thalassemia are: low levels of hemoglobin and red blood cells, with normal or increased iron levels, anisocytosis, poikilocytosis, increased levels of indirect bilirubin and the number of sideroblasts;
- the main clinical signs are pale skin, enlarged spleen, bone deformities and delayed physical development in children;
- The prognosis for the disease is generally favorable; symptomatic therapy is carried out.
Article rating
We have put a lot of effort into making sure you can read this article and would welcome your feedback in the form of a rating. The author will be pleased to see that you were interested in this material. Thank you!
(9 4,67 of 5) Loading...
If you liked the article, share it with your friends!
You might be interested
Source: https://UstamiVrachey.ru/hematologiya/talassemia
Treatment of thalassemia
Therapeutic measures depend on:
- The severity of erythropoiesis,
- The degree of damage to genes by the mutation process.
The main methods of treating thalassemia include:
- Diet. Its task is to reduce the absorption of Fe by the intestines. To do this, nuts, soy products, tea/cocoa are included in the menu.
- Introduction of chelates. These agents increase the activity of basic drugs and also eliminate excess Fe. Used daily.
- Receiving "Desferal". This drug binds Fe, thereby preventing its stagnation in tissues. The medicine does not affect hemoglobin levels.
- Transfusion of blood, red blood cells (filtered and thawed), red blood cells. The method is used in severe cases of the disease, when it is necessary to save the patient’s life.
- Taking large doses of glucocorticoids. They are appropriate when a hemolytic crisis develops.
- Splenectomy. The operation is prescribed for an enlarged spleen. Splenectomy is performed from the age of 5, but 8-10 years is considered an acceptable age. There is improvement after surgery, but there is a high risk of infection.
After surgery, a donor organ transplant is necessary: preferably, from one of the closest relatives.
Symptomatic drugs necessary for the treatment of thalassemia
- Hepatoprotectors,
- Ascorbic acid (removes excess Fe),
- Products based on folic acid,
- B vitamins.
It is impossible to completely recover from the disease.